

Oligodendrocyte Nf1 Controls Aberrant Notch Activation and Regulates Myelin Structure and Behavior
- PMID: 28423318
- PMCID: PMC5828008
- DOI: 10.1016/j.celrep.2017.03.073
Oligodendrocyte Nf1 Controls Aberrant Notch Activation and Regulates Myelin Structure and Behavior
Abstract
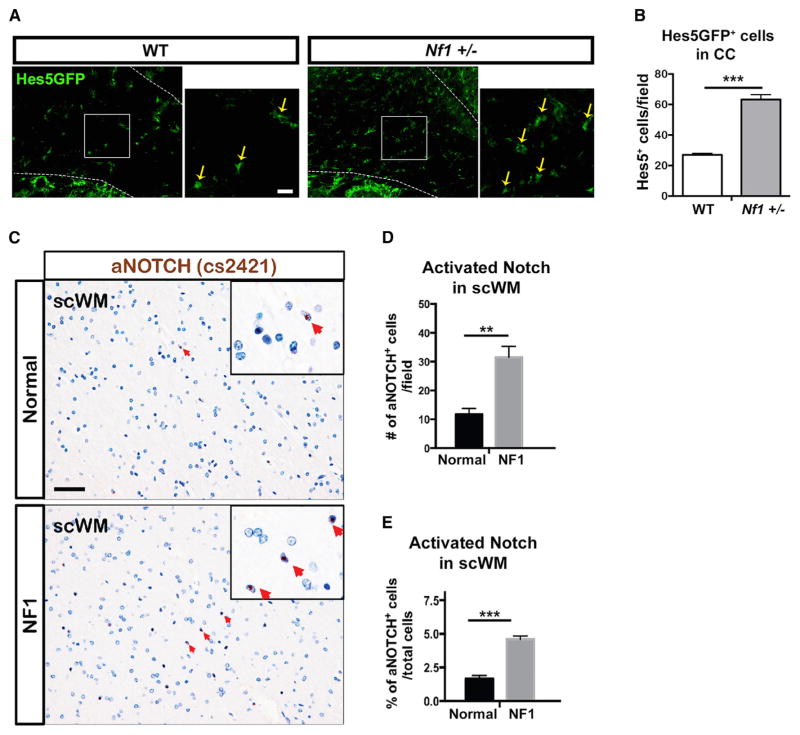
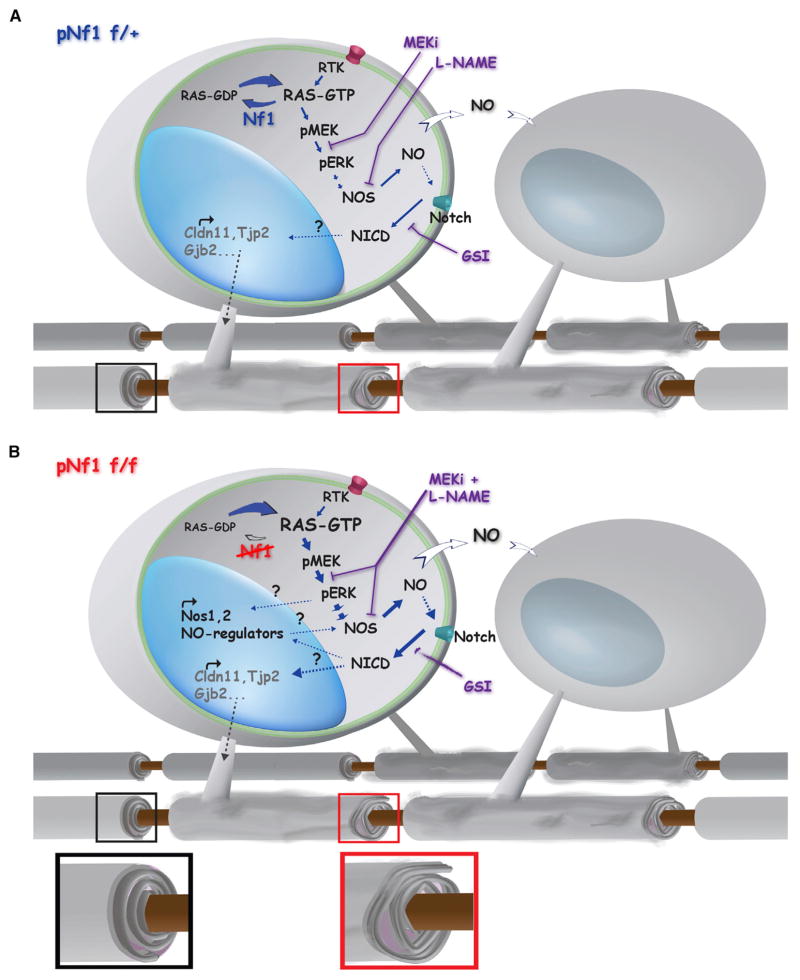
The RASopathy neurofibromatosis type 1 (NF1) is one of the most common autosomal dominant genetic disorders. In NF1 patients, neurological issues may result from damaged myelin, and mice with a neurofibromin gene (Nf1) mutation show white matter (WM) defects including myelin decompaction. Using mouse genetics, we find that altered Nf1 gene-dose in mature oligodendrocytes results in progressive myelin defects and behavioral abnormalities mediated by aberrant Notch activation. Blocking Notch, upstream mitogen-activated protein kinase (MAPK), or nitric oxide signaling rescues myelin defects in hemizygous Nf1 mutants, and pharmacological gamma secretase inhibition rescues aberrant behavior with no effects in wild-type (WT) mice. Concomitant pathway inhibition rescues myelin abnormalities in homozygous mutants. Notch activation is also observed in Nf1+/- mouse brains, and cells containing active Notch are increased in NF1 patient WM. We thus identify Notch as an Nf1 effector regulating myelin structure and behavior in a RASopathy and suggest that inhibition of Notch signaling may be a therapeutic strategy for NF1.
Keywords: glia; rasopathy.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Oligodendrocyte RasG12V expressed in its endogenous locus disrupts myelin structure through increased MAPK, nitric oxide, and notch signaling.Glia. 2017 Dec;65(12):1990-2002. doi: 10.1002/glia.23209. Epub 2017 Aug 30. Glia. 2017. PMID: 28856719 Free PMC article.
-
Nf1 loss and Ras hyperactivation in oligodendrocytes induce NOS-driven defects in myelin and vasculature.Cell Rep. 2013 Sep 26;4(6):1197-212. doi: 10.1016/j.celrep.2013.08.011. Epub 2013 Sep 12. Cell Rep. 2013. PMID: 24035394 Free PMC article.
-
Brain-wide structural and functional disruption in mice with oligodendrocyte-specific Nf1 deletion is rescued by inhibition of nitric oxide synthase.Proc Natl Acad Sci U S A. 2020 Sep 8;117(36):22506-22513. doi: 10.1073/pnas.2008391117. Epub 2020 Aug 24. Proc Natl Acad Sci U S A. 2020. PMID: 32839340 Free PMC article.
-
Cross-talk between F3/contactin and Notch at axoglial interface: a role in oligodendrocyte development.Dev Neurosci. 2006;28(1-2):25-33. doi: 10.1159/000090750. Dev Neurosci. 2006. PMID: 16508301 Review.
-
Neurofibromin in neurofibromatosis type 1 - mutations in NF1gene as a cause of disease.Dev Period Med. 2014 Jul-Sep;18(3):297-306. Dev Period Med. 2014. PMID: 25182393 Review.
Cited by
-
In vivo Functional Genomics for Undiagnosed Patients: The Impact of Small GTPases Signaling Dysregulation at Pan-Embryo Developmental Scale.Front Cell Dev Biol. 2021 May 25;9:642235. doi: 10.3389/fcell.2021.642235. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34124035 Free PMC article. Review.
-
Daam2 Regulates Myelin Structure and the Oligodendrocyte Actin Cytoskeleton through Rac1 and Gelsolin.J Neurosci. 2022 Mar 2;42(9):1679-1691. doi: 10.1523/JNEUROSCI.1517-21.2022. Epub 2022 Jan 31. J Neurosci. 2022. PMID: 35101966 Free PMC article.
-
Granulins Regulate Aging Kinetics in the Adult Zebrafish Telencephalon.Cells. 2020 Feb 3;9(2):350. doi: 10.3390/cells9020350. Cells. 2020. PMID: 32028681 Free PMC article.
-
A new era for myelin research in Neurofibromatosis type 1.Glia. 2023 Dec;71(12):2701-2719. doi: 10.1002/glia.24432. Epub 2023 Jun 29. Glia. 2023. PMID: 37382486 Free PMC article. Review.
-
Autism-associated Nf1 deficiency disrupts corticocortical and corticostriatal functional connectivity in human and mouse.Neurobiol Dis. 2019 Oct;130:104479. doi: 10.1016/j.nbd.2019.104479. Epub 2019 May 22. Neurobiol Dis. 2019. PMID: 31128207 Free PMC article.
References
-
- Acosta MT, Gioia GA, Silva AJ. Neurofibromatosis type 1: new insights into neurocognitive issues. Curr Neurol Neurosci Rep. 2006;6:136–143. - PubMed
-
- Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR, et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett. 2008;18:6501–6504. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
