

Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2
- PMID: 28424246
- PMCID: PMC5413324
- DOI: 10.1084/jem.20160894
Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2
Abstract
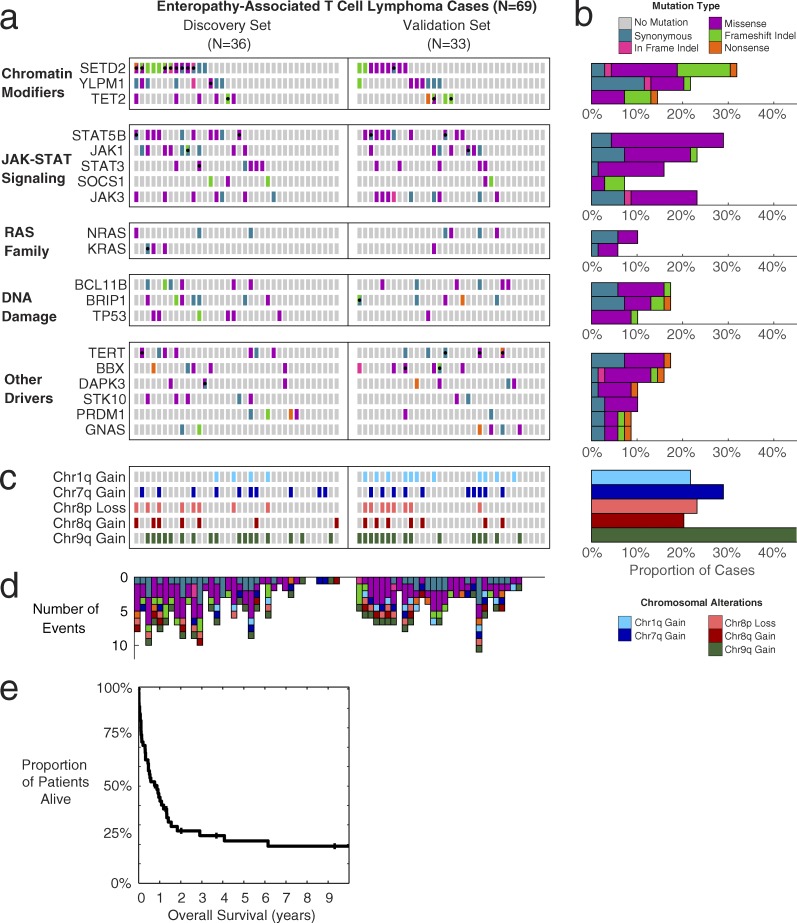
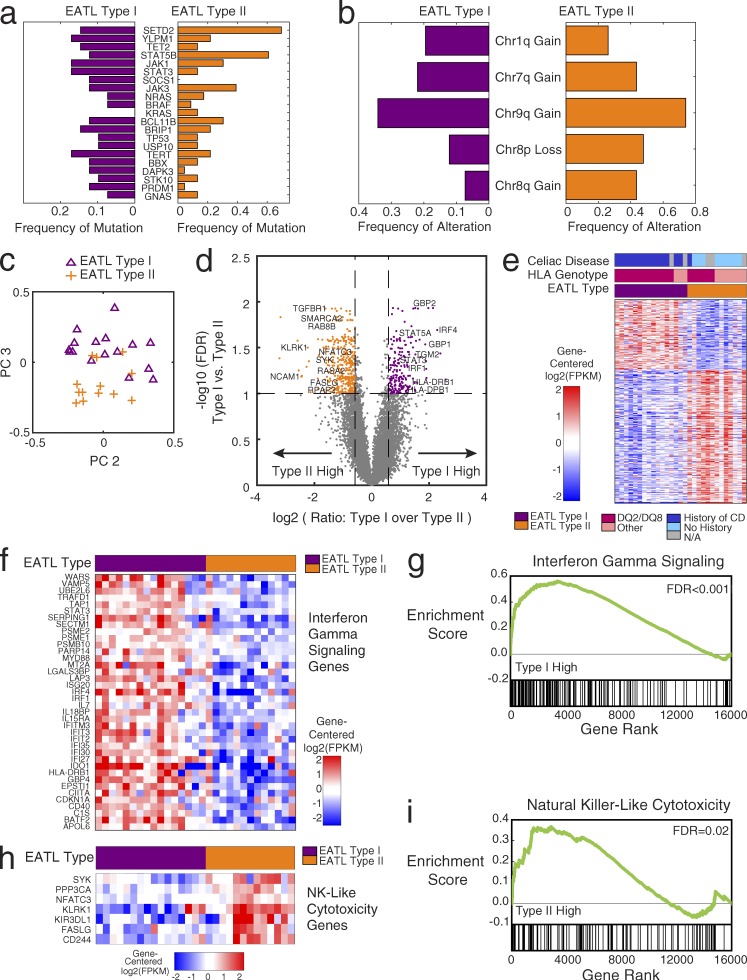
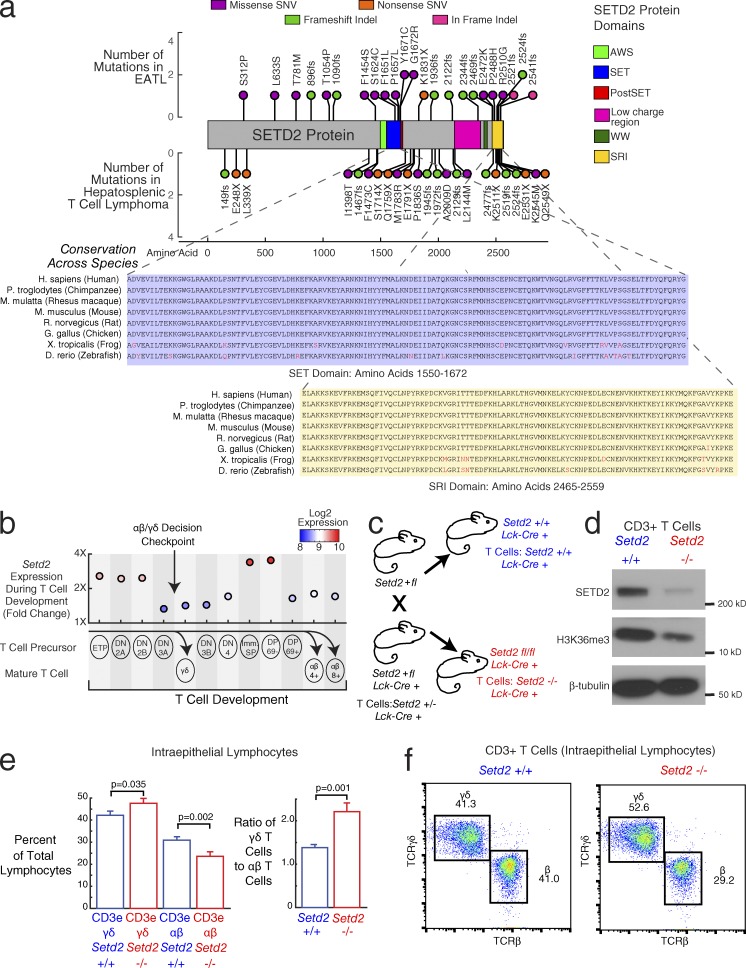
Enteropathy-associated T cell lymphoma (EATL) is a lethal, and the most common, neoplastic complication of celiac disease. Here, we defined the genetic landscape of EATL through whole-exome sequencing of 69 EATL tumors. SETD2 was the most frequently silenced gene in EATL (32% of cases). The JAK-STAT pathway was the most frequently mutated pathway, with frequent mutations in STAT5B as well as JAK1, JAK3, STAT3, and SOCS1 We also identified mutations in KRAS, TP53, and TERT Type I EATL and type II EATL (monomorphic epitheliotropic intestinal T cell lymphoma) had highly overlapping genetic alterations indicating shared mechanisms underlying their pathogenesis. We modeled the effects of SETD2 loss in vivo by developing a T cell-specific knockout mouse. These mice manifested an expansion of γδ T cells, indicating novel roles for SETD2 in T cell development and lymphomagenesis. Our data render the most comprehensive genetic portrait yet of this uncommon but lethal disease and may inform future classification schemes.
© 2017 Moffitt et al.
Figures
Similar articles
-
Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations.Nat Commun. 2016 Sep 7;7:12602. doi: 10.1038/ncomms12602. Nat Commun. 2016. PMID: 27600764 Free PMC article.
-
Immunophenotypic Spectrum and Genomic Landscape of Refractory Celiac Disease Type II.Am J Surg Pathol. 2021 Jul 1;45(7):905-916. doi: 10.1097/PAS.0000000000001658. Am J Surg Pathol. 2021. PMID: 33544565
-
[Monomorphic epitheliotropic intestinal T-cell lymphoma: a clinicopathological analysis of twelve cases].Zhonghua Bing Li Xue Za Zhi. 2020 Jan 8;49(1):17-21. doi: 10.3760/cma.j.issn.0529-5807.2020.01.004. Zhonghua Bing Li Xue Za Zhi. 2020. PMID: 31914529 Chinese.
-
Pathogenesis of Enteropathy-Associated T Cell Lymphoma.Curr Hematol Malig Rep. 2018 Aug;13(4):308-317. doi: 10.1007/s11899-018-0459-5. Curr Hematol Malig Rep. 2018. PMID: 29943210 Review.
-
Enteropathy-Associated T cell Lymphoma.Curr Hematol Malig Rep. 2021 Apr;16(2):140-147. doi: 10.1007/s11899-021-00634-4. Epub 2021 May 19. Curr Hematol Malig Rep. 2021. PMID: 34009525 Review.
Cited by
-
Monomorphic epitheliotropic intestinal T-cell lymphoma: report of four cases and literature review.J Int Med Res. 2024 Aug;52(8):3000605241271756. doi: 10.1177/03000605241271756. J Int Med Res. 2024. PMID: 39197860 Free PMC article. Review.
-
Genetic and phenotypic characterization of indolent T-cell lymphoproliferative disorders of the gastrointestinal tract.Haematologica. 2020 Jul;105(7):1895-1906. doi: 10.3324/haematol.2019.230961. Epub 2019 Sep 26. Haematologica. 2020. PMID: 31558678 Free PMC article.
-
Clinicopathological and molecular genomic features of monomorphic epitheliotropic intestinal T-cell lymphoma in the Chinese population: a study of 20 cases.Diagn Pathol. 2021 Dec 12;16(1):114. doi: 10.1186/s13000-021-01173-5. Diagn Pathol. 2021. PMID: 34895266 Free PMC article.
-
Janus Kinase Signaling: Oncogenic Criminal of Lymphoid Cancers.Cancers (Basel). 2021 Oct 14;13(20):5147. doi: 10.3390/cancers13205147. Cancers (Basel). 2021. PMID: 34680295 Free PMC article. Review.
-
Molecular Insights Into Pathogenesis of Peripheral T Cell Lymphoma: a Review.Curr Hematol Malig Rep. 2018 Aug;13(4):318-328. doi: 10.1007/s11899-018-0460-z. Curr Hematol Malig Rep. 2018. PMID: 29951889 Review.
References
-
- Belhadj K., Reyes F., Farcet J.-P., Tilly H., Bastard C., Angonin R., Deconinck E., Charlotte F., Leblond V., Labouyrie E., et al. . 2003. Hepatosplenic γδ T-cell lymphoma is a rare clinicopathologic entity with poor outcome: Report on a series of 21 patients. Blood. 102:4261–4269. 10.1182/blood-2003-05-1675 - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
