

Improving genetic diagnosis in Mendelian disease with transcriptome sequencing
- PMID: 28424332
- PMCID: PMC5548421
- DOI: 10.1126/scitranslmed.aal5209
Improving genetic diagnosis in Mendelian disease with transcriptome sequencing
Abstract
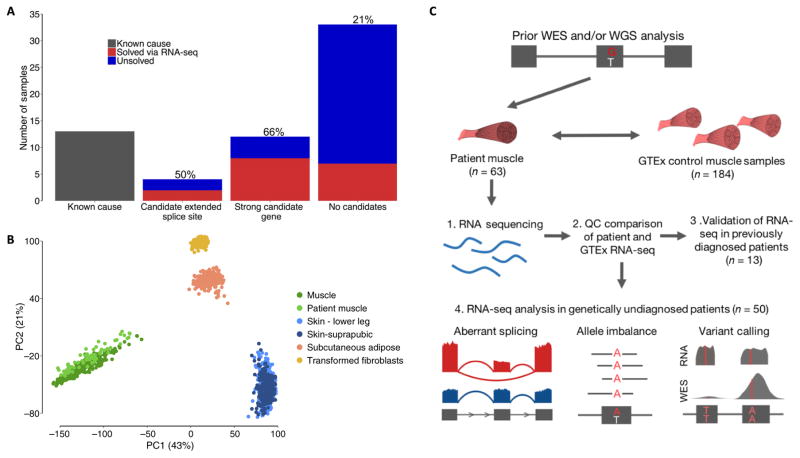
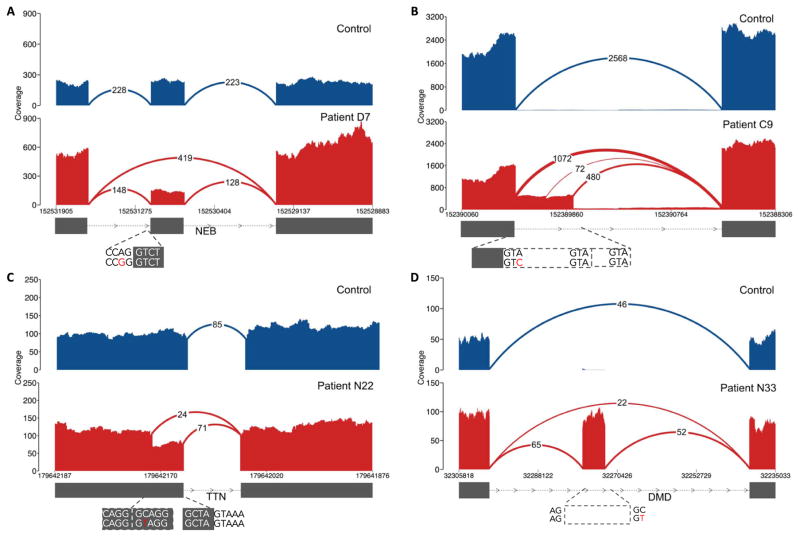
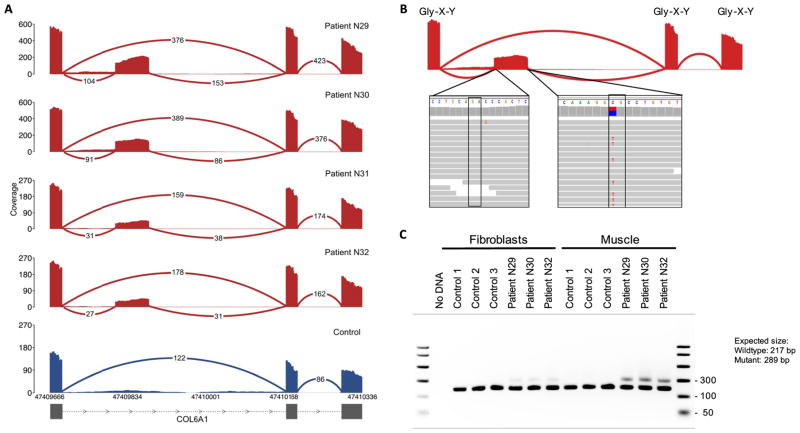
Exome and whole-genome sequencing are becoming increasingly routine approaches in Mendelian disease diagnosis. Despite their success, the current diagnostic rate for genomic analyses across a variety of rare diseases is approximately 25 to 50%. We explore the utility of transcriptome sequencing [RNA sequencing (RNA-seq)] as a complementary diagnostic tool in a cohort of 50 patients with genetically undiagnosed rare muscle disorders. We describe an integrated approach to analyze patient muscle RNA-seq, leveraging an analysis framework focused on the detection of transcript-level changes that are unique to the patient compared to more than 180 control skeletal muscle samples. We demonstrate the power of RNA-seq to validate candidate splice-disrupting mutations and to identify splice-altering variants in both exonic and deep intronic regions, yielding an overall diagnosis rate of 35%. We also report the discovery of a highly recurrent de novo intronic mutation in COL6A1 that results in a dominantly acting splice-gain event, disrupting the critical glycine repeat motif of the triple helical domain. We identify this pathogenic variant in a total of 27 genetically unsolved patients in an external collagen VI-like dystrophy cohort, thus explaining approximately 25% of patients clinically suggestive of having collagen VI dystrophy in whom prior genetic analysis is negative. Overall, this study represents a large systematic application of transcriptome sequencing to rare disease diagnosis and highlights its utility for the detection and interpretation of variants missed by current standard diagnostic approaches.
Copyright © 2017, American Association for the Advancement of Science.
Figures
Comment in
-
Genetic testing: The diagnostic power of RNA-seq.Nat Rev Genet. 2017 Jul;18(7):392-393. doi: 10.1038/nrg.2017.39. Epub 2017 May 8. Nat Rev Genet. 2017. PMID: 28479597 No abstract available.
-
Clinical Utility of Transcriptome Sequencing: Toward a Better Diagnosis for Mendelian Disorders.Clin Chem. 2018 Jun;64(6):882-884. doi: 10.1373/clinchem.2017.276980. Epub 2017 Nov 2. Clin Chem. 2018. PMID: 29097506 No abstract available.
-
Increasing diagnostic yield by RNA-Sequencing in rare disease-bypass hurdles of interpreting intronic or splice-altering variants.Ann Transl Med. 2018 Apr;6(7):126. doi: 10.21037/atm.2018.01.14. Ann Transl Med. 2018. PMID: 29955586 Free PMC article. No abstract available.
References
-
- Ankala A, da Silva C, Gualandi F, Ferlini A, Bean LJ, Collins C, Tanner AK, Hegde MR. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015;77:206–214. - PubMed
-
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. - PMC - PubMed
-
- Taylor JC, Martin HC, Lise S, Broxholme J, Cazier J-B, Rimmer A, Kanapin A, Lunter G, Fiddy S, Allan C, Aricescu AR, Attar M, Babbs C, Becq J, Beeson D, Bento C, Bignell P, Blair E, Buckle VJ, Bull K, Cais O, Cario H, Chapel H, Copley RR, Cornall R, Craft J, Dahan K, Davenport EE, Dendrou C, Devuyst O, Fenwick AL, Flint J, Fugger L, Gilbert RD, Goriely A, Green A, Greger IH, Grocock R, Gruszczyk AV, Hastings R, Hatton E, Higgs D, Hill A, Holmes C, Howard M, Hughes L, Humburg P, Johnson D, Karpe F, Kingsbury Z, Kini U, Knight JC, Krohn J, Lamble S, Langman C, Lonie L, Luck J, McCarthy D, McGowan SJ, McMullin MF, Miller KA, Murray L, Németh AH, Andrew MN, Nutt D, Ormondroyd E, Bang Oturai A, Pagnamenta A, Patel SY, Percy M, Petousi N, Piazza P, Piret SE, Polanco-Echeverry G, Popitsch N, Powrie F, Pugh C, Quek L, Robbins PA, Robson K, Russo A, Sahgal N, van Schouwenburg PA, Schuh A, Silverman E, Simmons A, Sørensen PS, Sweeney E, Taylor J, Thakker RV, Tomlinson I, Trebes A, Twigg SRF, Uhlig HH, Vyas P, Vyse T, Wall SA, Watkins H, Whyte MP, Witty L, Wright B, Yau C, Buck D, Humphray S, Ratcliffe PJ, Bell JI, Wilkie AOM, Bentley D, Donnelly P, McVean G. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47:717–726. - PMC - PubMed
-
- Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, Harrell TM, McMillin MJ, Wiszniewski W, Gambin T, Coban Akdemir ZH, Doheny K, Scott AF, Avramopoulos D, Chakravarti A, Hoover-Fong J, Mathews D, Witmer PD, Ling H, Hetrick K, Watkins L, Patterson KE, Reinier F, Blue E, Muzny D, Kircher M, Bilguvar K, López-Giráldez F, Sutton VR, Tabor HK, Leal SM, Gunel M, Mane S, Gibbs RA, Boerwinkle E, Hamosh A, Shendure J, Lupski JR, Lifton RP, Valle D, Nickerson DA, Bamshad MJ Centers for Mendelian Genomics. The genetic basis of Mendelian phenotypes: Discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97:199–215. - PMC - PubMed
-
- MacArthur D, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, Conrad DF, Cooper GM, Cox NJ, Daly MJ, Gerstein MB, Goldstein DB, Hirschhorn JN, Leal SM, Pennacchio LA, Stamatoyannopoulos JA, Sunyaev SR, Valle D, Voight BF, Winckler W, Gunter C. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- R01 DA006227/DA/NIDA NIH HHS/United States
- R01 AR044345/AR/NIAMS NIH HHS/United States
- R01 HD075802/HD/NICHD NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- F32 GM115208/GM/NIGMS NIH HHS/United States
- T32 GM007748/GM/NIGMS NIH HHS/United States
- R01 NS080929/NS/NINDS NIH HHS/United States
- T32 GM096911/GM/NIGMS NIH HHS/United States
- U54 HD090255/HD/NICHD NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 GM104371/GM/NIGMS NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- L60 MD010032/MD/NIMHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
