

Exhaustive search of linear information encoding protein-peptide recognition
- PMID: 28426660
- PMCID: PMC5417721
- DOI: 10.1371/journal.pcbi.1005499
Exhaustive search of linear information encoding protein-peptide recognition
Abstract
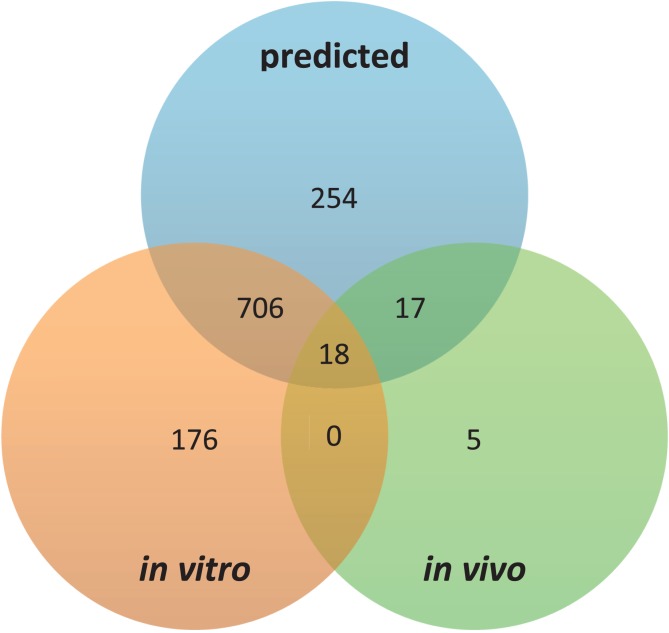
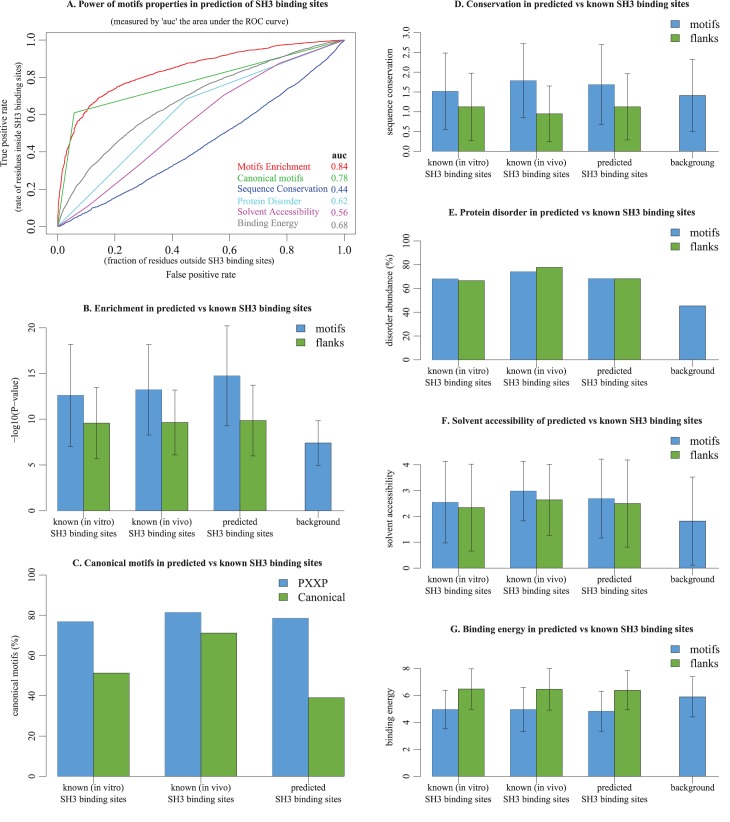
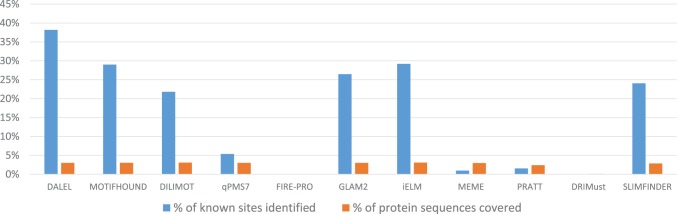
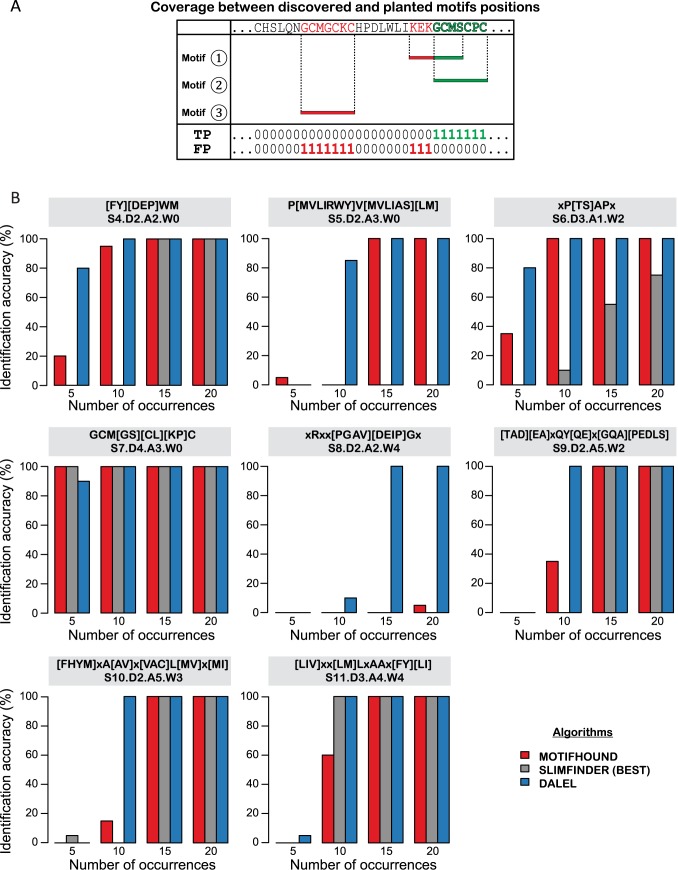
High-throughput in vitro methods have been extensively applied to identify linear information that encodes peptide recognition. However, these methods are limited in number of peptides, sequence variation, and length of peptides that can be explored, and often produce solutions that are not found in the cell. Despite the large number of methods developed to attempt addressing these issues, the exhaustive search of linear information encoding protein-peptide recognition has been so far physically unfeasible. Here, we describe a strategy, called DALEL, for the exhaustive search of linear sequence information encoded in proteins that bind to a common partner. We applied DALEL to explore binding specificity of SH3 domains in the budding yeast Saccharomyces cerevisiae. Using only the polypeptide sequences of SH3 domain binding proteins, we succeeded in identifying the majority of known SH3 binding sites previously discovered either in vitro or in vivo. Moreover, we discovered a number of sites with both non-canonical sequences and distinct properties that may serve ancillary roles in peptide recognition. We compared DALEL to a variety of state-of-the-art algorithms in the blind identification of known binding sites of the human Grb2 SH3 domain. We also benchmarked DALEL on curated biological motifs derived from the ELM database to evaluate the effect of increasing/decreasing the enrichment of the motifs. Our strategy can be applied in conjunction with experimental data of proteins interacting with a common partner to identify binding sites among them. Yet, our strategy can also be applied to any group of proteins of interest to identify enriched linear motifs or to exhaustively explore the space of linear information encoded in a polypeptide sequence. Finally, we have developed a webserver located at http://michnick.bcm.umontreal.ca/dalel, offering user-friendly interface and providing different scenarios utilizing DALEL.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
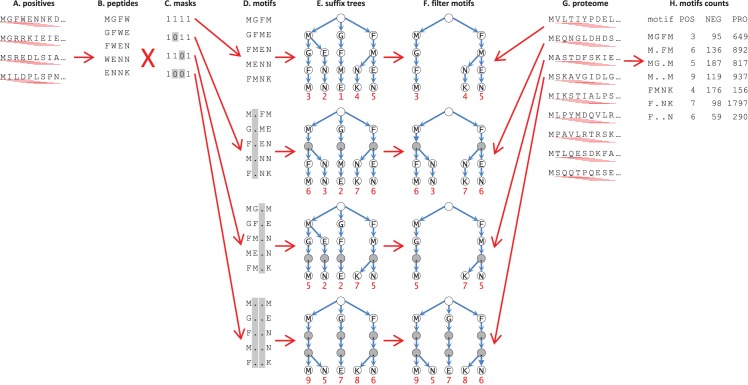
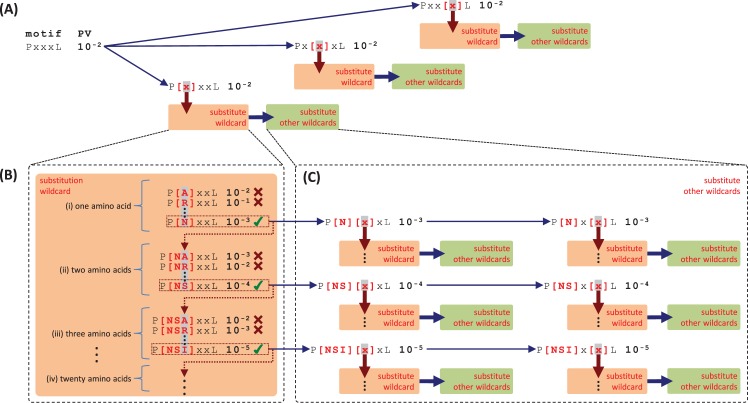
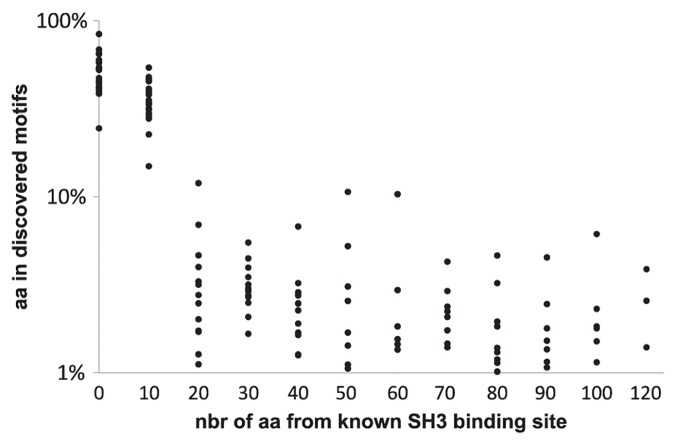
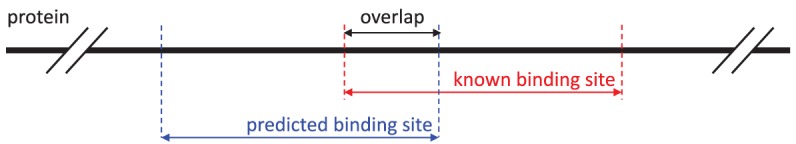
References
-
- Tong AH, Drees B, Nardelli G, Bader GD, Brannetti B, Castagnoli L, et al. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science. 2002;295(5553):321–4. doi: 10.1126/science.1064987 - DOI - PubMed
-
- Dinkel H, Van Roey K, Michael S, Davey NE, Weatheritt RJ, Born D, et al. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 2014;42(Database issue):D259–66. PubMed Central PMCID: PMCPMC3964949. doi: 10.1093/nar/gkt1047 - DOI - PMC - PubMed
-
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–30. PubMed Central PMCID: PMC3965110. doi: 10.1093/nar/gkt1223 - DOI - PMC - PubMed
-
- Tonikian R, Xin X, Toret CP, Gfeller D, Landgraf C, Panni S, et al. Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLoS Biol. 2009;7(10):e1000218 PubMed Central PMCID: PMC2756588. doi: 10.1371/journal.pbio.1000218 - DOI - PMC - PubMed
-
- Watterson S, Ghazal P. Use of logic theory in understanding regulatory pathway signaling in response to infection. Future Microbiol. 2010;5(2):163–76. doi: 10.2217/fmb.10.8 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
