

Bisphenol A activates EGFR and ERK promoting proliferation, tumor spheroid formation and resistance to EGFR pathway inhibition in estrogen receptor-negative inflammatory breast cancer cells
- PMID: 28426875
- PMCID: PMC5963742
- DOI: 10.1093/carcin/bgx003
Bisphenol A activates EGFR and ERK promoting proliferation, tumor spheroid formation and resistance to EGFR pathway inhibition in estrogen receptor-negative inflammatory breast cancer cells
Abstract
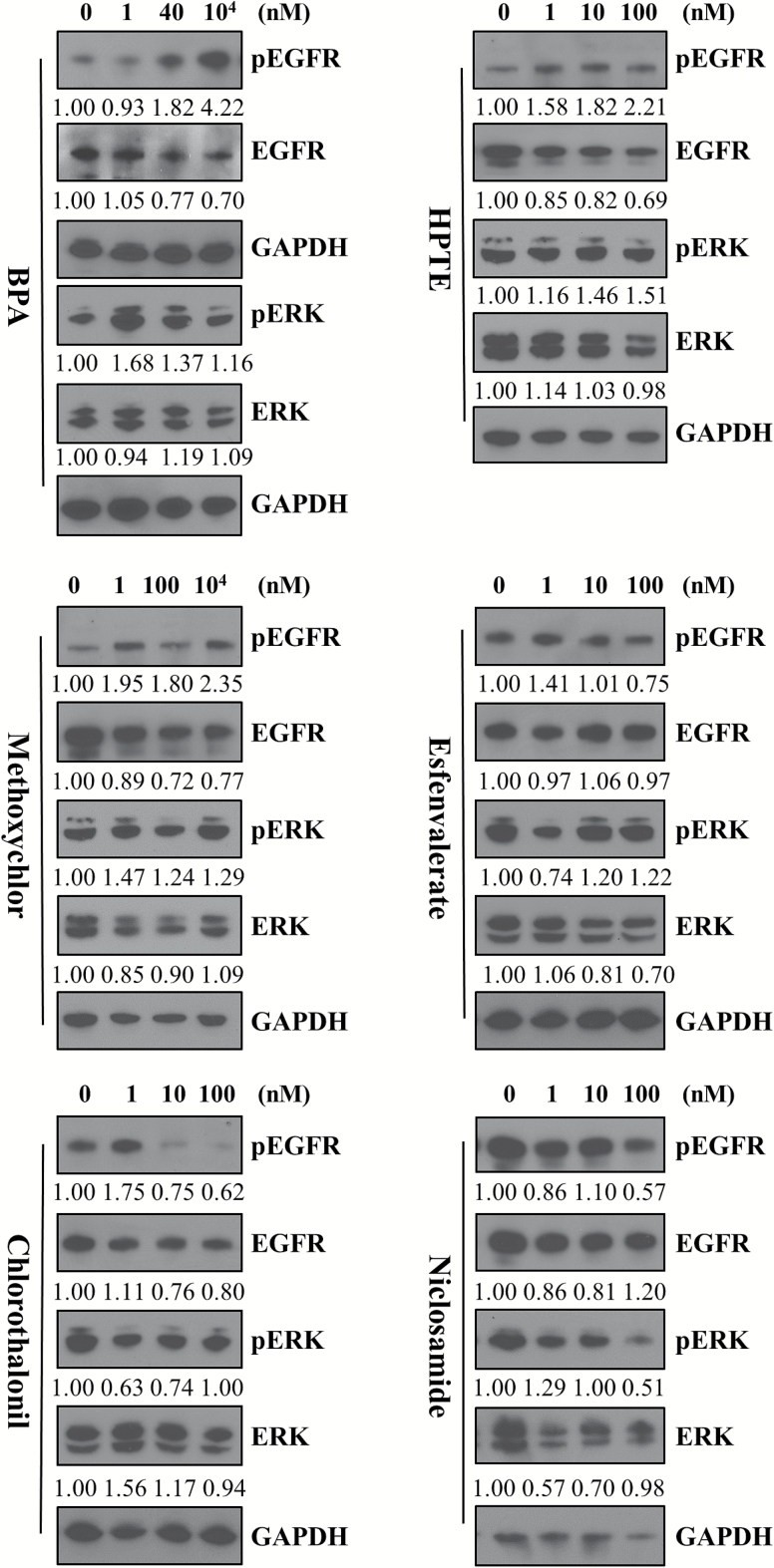
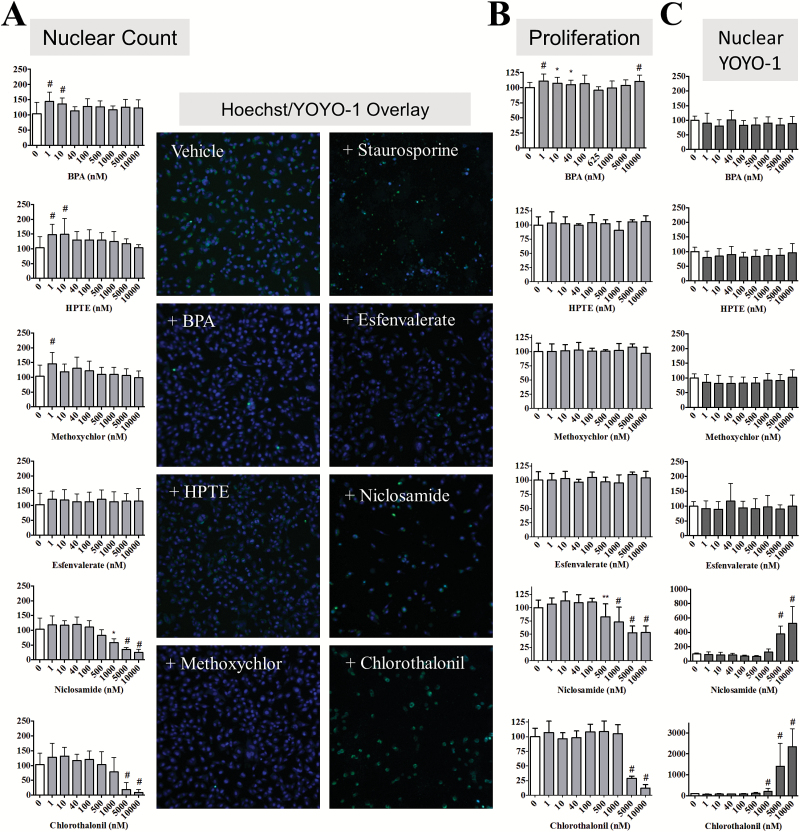
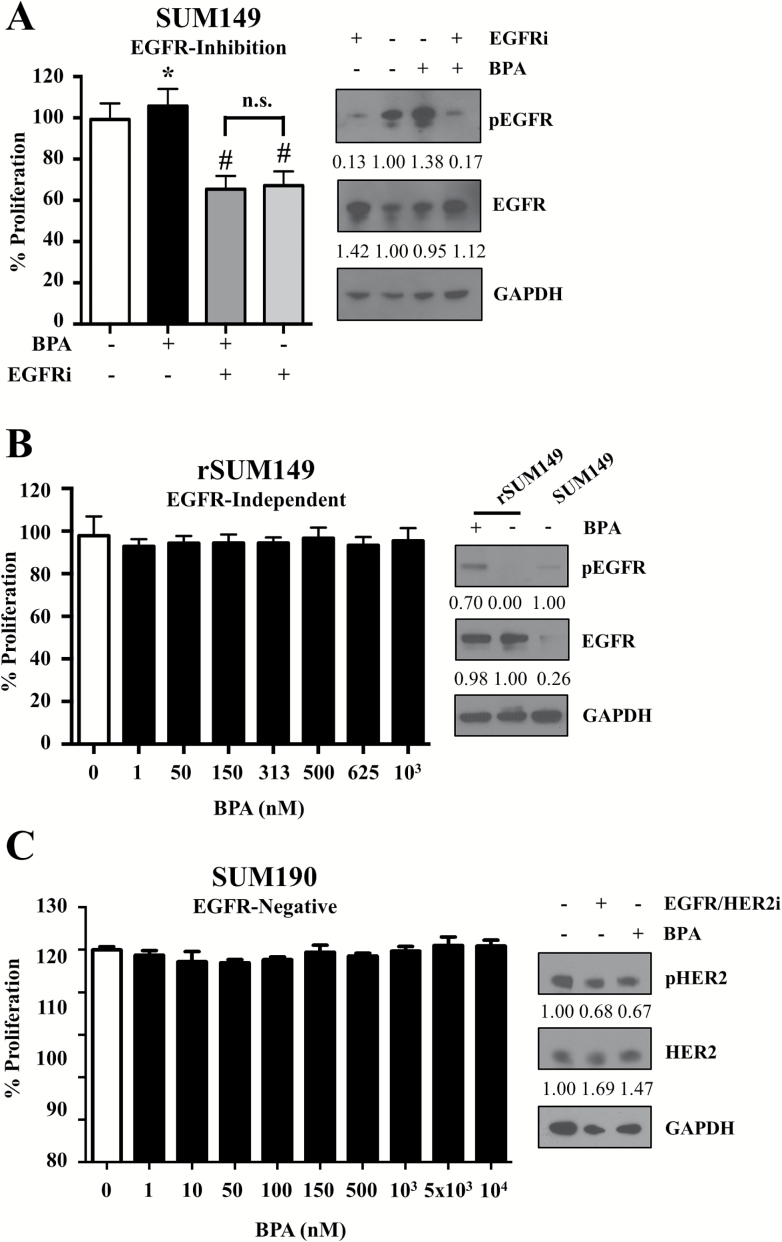
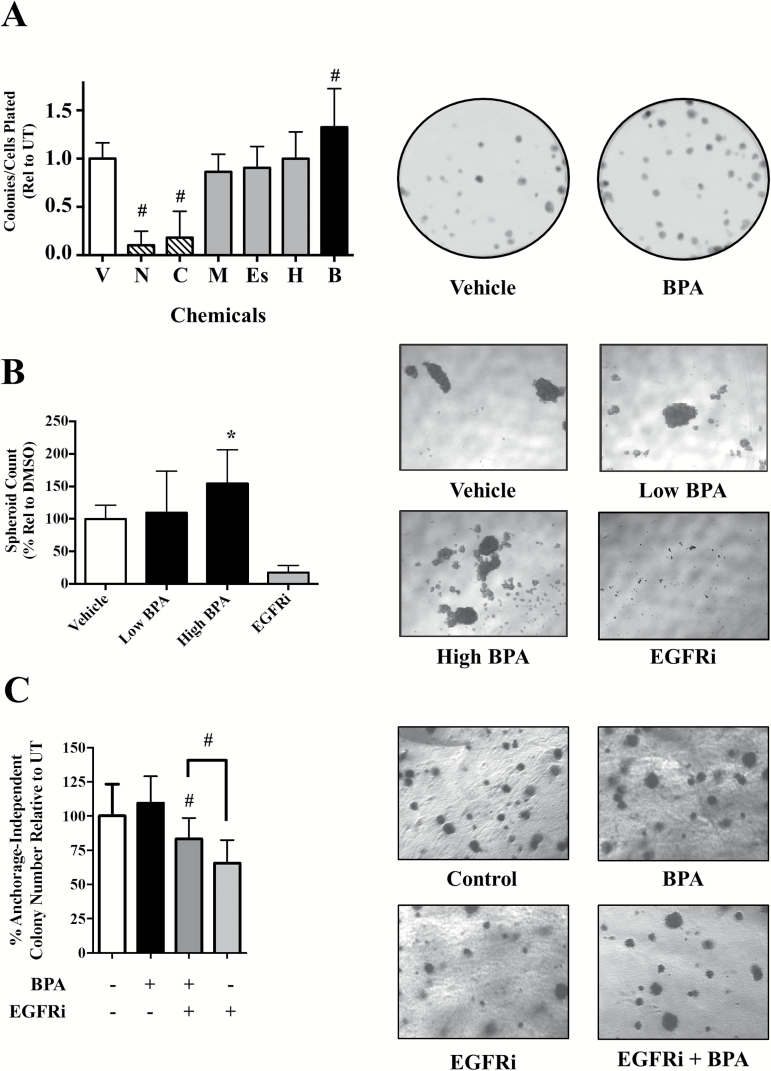
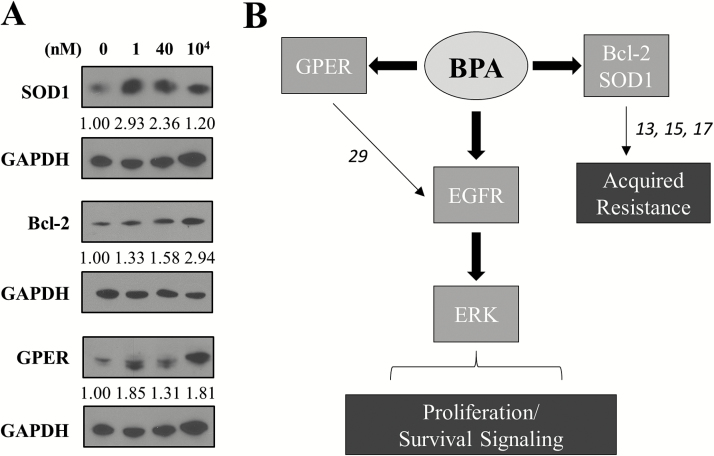
Emerging evidence from epidemiological studies suggests a link between environmental chemical exposure and progression of aggressive breast cancer subtypes. Of all clinically distinct types of breast cancers, the most lethal phenotypic variant is inflammatory breast cancer (IBC). Overexpression of epidermal growth factor receptors (EGFR/HER2) along with estrogen receptor (ER) negativity is common in IBC tumor cells, which instead of a solid mass present as rapidly proliferating diffuse tumor cell clusters. Our previous studies have demonstrated a role of an adaptive response of increased antioxidants in acquired resistance to EGFR-targeting drugs in IBC. Environmental chemicals are known to induce oxidative stress resulting in perturbations in signal transduction pathways. It is therefore of interest to identify chemicals that can potentiate EGFR mitogenic effects in IBC. Herein, we assessed in ER-negative IBC cells a subset of chemicals from the EPA ToxCast set for their effect on EGFR activation and in multiple cancer phenotypic assays. We demonstrated that endocrine-disrupting chemicals such as bisphenol A (BPA) and 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane can increase EGFR/ERK signaling. BPA also caused a corresponding increase in expression of SOD1 and anti-apoptotic Bcl-2, key markers of antioxidant and anti-apoptotic processes. BPA potentiated clonogenic growth and tumor spheroid formation in vitro, reflecting IBC-specific pathological characteristics. Furthermore, we identified that BPA was able to attenuate the inhibitory effect of an EGFR targeted drug in a longer-term anchorage-independent growth assay. These findings provide a potential mechanistic basis for environmental chemicals such as BPA in potentiating a hyperproliferative and death-resistant phenotype in cancer cells by activating mitogenic pathways to which the tumor cells are addicted for survival.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Comment in
-
Endocrine Disruptors: Adverse Health Effects Mediated by EGFR?Trends Endocrinol Metab. 2018 Feb;29(2):69-71. doi: 10.1016/j.tem.2017.12.003. Epub 2017 Dec 29. Trends Endocrinol Metab. 2018. PMID: 29292062
Similar articles
-
Involvement of activating ERK1/2 through G protein coupled receptor 30 and estrogen receptor α/β in low doses of bisphenol A promoting growth of Sertoli TM4 cells.Toxicol Lett. 2014 Apr 7;226(1):81-9. doi: 10.1016/j.toxlet.2014.01.035. Epub 2014 Feb 2. Toxicol Lett. 2014. PMID: 24495410
-
Low concentrations of bisphenol A induce mouse spermatogonial cell proliferation by G protein-coupled receptor 30 and estrogen receptor-α.Environ Health Perspect. 2011 Dec;119(12):1775-80. doi: 10.1289/ehp.1103781. Epub 2011 Aug 3. Environ Health Perspect. 2011. PMID: 21813366 Free PMC article.
-
Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways.Mol Cancer. 2017 Mar 7;16(1):57. doi: 10.1186/s12943-017-0621-z. Mol Cancer. 2017. PMID: 28270211 Free PMC article.
-
Stop eating plastic, molecular signaling of bisphenol A in breast cancer.Environ Sci Pollut Res Int. 2018 Aug;25(24):23624-23630. doi: 10.1007/s11356-018-2540-y. Epub 2018 Jun 29. Environ Sci Pollut Res Int. 2018. PMID: 29959737 Review.
-
Role of epidermal growth factor receptor in breast cancer.Breast Cancer Res Treat. 2012 Nov;136(2):331-45. doi: 10.1007/s10549-012-2289-9. Epub 2012 Oct 17. Breast Cancer Res Treat. 2012. PMID: 23073759 Free PMC article. Review.
Cited by
-
Pharmacological targeting of GLI1 inhibits proliferation, tumor emboli formation and in vivo tumor growth of inflammatory breast cancer cells.Cancer Lett. 2017 Dec 28;411:136-149. doi: 10.1016/j.canlet.2017.09.033. Epub 2017 Sep 28. Cancer Lett. 2017. PMID: 28965853 Free PMC article.
-
Bisphenol-A and Nonylphenol Induce Apoptosis in Reproductive Tract Cancer Cell Lines by the Activation of ADAM17.Int J Mol Sci. 2018 Jul 31;19(8):2238. doi: 10.3390/ijms19082238. Int J Mol Sci. 2018. PMID: 30065191 Free PMC article.
-
ERK is a Pivotal Player of Chemo-Immune-Resistance in Cancer.Int J Mol Sci. 2019 May 21;20(10):2505. doi: 10.3390/ijms20102505. Int J Mol Sci. 2019. PMID: 31117237 Free PMC article. Review.
-
Re-evaluation of the risks to public health related to the presence of bisphenol A (BPA) in foodstuffs.EFSA J. 2023 Apr 19;21(4):e06857. doi: 10.2903/j.efsa.2023.6857. eCollection 2023 Apr. EFSA J. 2023. PMID: 37089179 Free PMC article.
-
Chemical mixture that targets the epidermal growth factor pathway impairs human trophoblast cell functions.Toxicol Appl Pharmacol. 2024 Feb;483:116804. doi: 10.1016/j.taap.2024.116804. Epub 2024 Jan 6. Toxicol Appl Pharmacol. 2024. PMID: 38185387 Free PMC article.
References
-
- Woodward W.A. (2015) Inflammatory breast cancer: unique biological and therapeutic considerations. Lancet. Oncol., 16, e568–e576. - PubMed
-
- Wingo P.A., et al. (2004) Population-based statistics for women diagnosed with inflammatory breast cancer (United States). Cancer Causes Control, 15, 321–328. - PubMed
-
- Boussen H., et al. (2008) Inflammatory breast cancer in Tunisia: reassessment of incidence and clinicopathological features. Semin. Oncol., 35, 17–24. - PubMed
-
- Duke T.J., et al. (2010) A cluster of inflammatory breast cancer (IBC) in an office setting: additional evidence of the importance of environmental factors in IBC etiology. Oncol. Rep., 24, 1277–1284. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
