

Hinge-deleted IgG4 blocker therapy for acetylcholine receptor myasthenia gravis in rhesus monkeys
- PMID: 28428630
- PMCID: PMC5430546
- DOI: 10.1038/s41598-017-01019-5
Hinge-deleted IgG4 blocker therapy for acetylcholine receptor myasthenia gravis in rhesus monkeys
Abstract
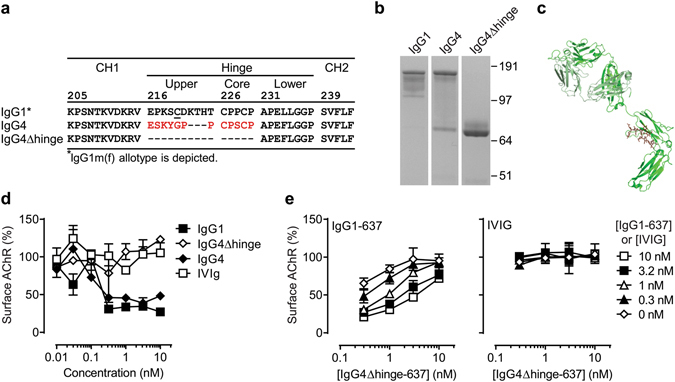
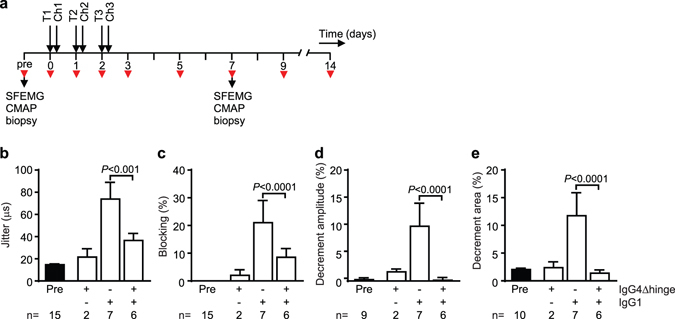
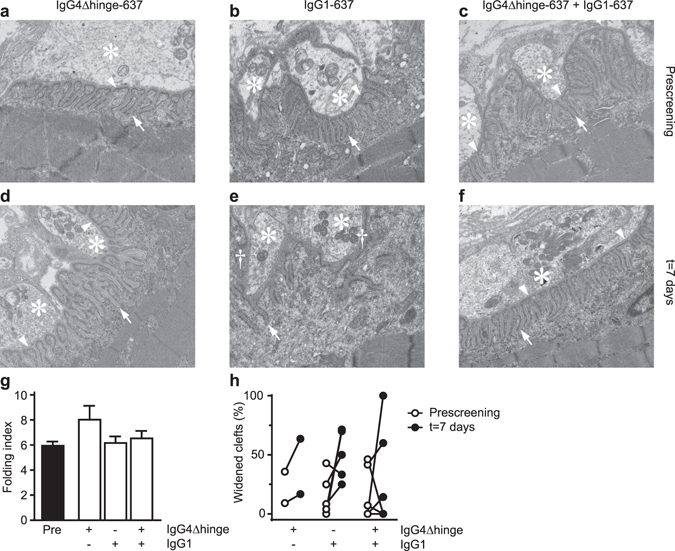
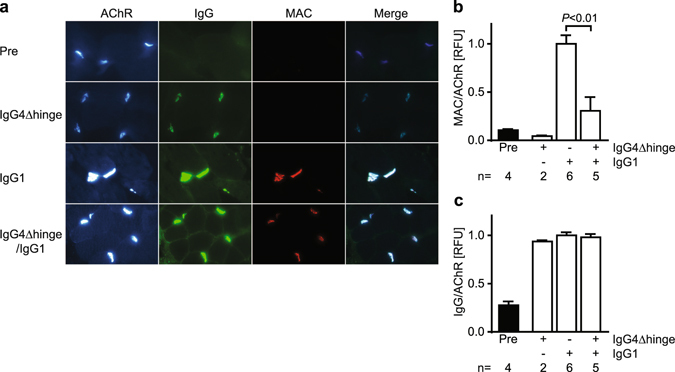
Autoantibodies against ion channels are the cause of numerous neurologic autoimmune disorders. Frequently, such pathogenic autoantibodies have a restricted epitope-specificity. In such cases, competing antibody formats devoid of pathogenic effector functions (blocker antibodies) have the potential to treat disease by displacing autoantibodies from their target. Here, we have used a model of the neuromuscular autoimmune disease myasthenia gravis in rhesus monkeys (Macaca mulatta) to test the therapeutic potential of a new blocker antibody: MG was induced by passive transfer of pathogenic acetylcholine receptor-specific monoclonal antibody IgG1-637. The effect of the blocker antibody (IgG4Δhinge-637, the hinge-deleted IgG4 version of IgG1-637) was assessed using decrement measurements and single-fiber electromyography. Three daily doses of 1.7 mg/kg IgG1-637 (cumulative dose 5 mg/kg) induced impairment of neuromuscular transmission, as demonstrated by significantly increased jitter, synaptic transmission failures (blockings) and a decrease in the amplitude of the compound muscle action potentials during repeated stimulations (decrement), without showing overt symptoms of muscle weakness. Treatment with three daily doses of 10 mg/kg IgG4Δhinge-637 significantly reduced the IgG1-637-induced increase in jitter, blockings and decrement. Together, these results represent proof-of principle data for therapy of acetylcholine receptor-myasthenia gravis with a monovalent antibody format that blocks binding of pathogenic autoantibodies.
Conflict of interest statement
These authors have a financial interest in Genmab: A.F.L., M.J., F.J.B., T.V., J.S. and P.W.H.I.P. have stock and/or warrants. M.L., P.M-M., J.S., M.H.d.B., and P.W.H.I.P own patent rights to the concept presented in this study. A.F.L., T.V., J.S., F.J.B., and P.W.H.I.P own patent rights to the IgG4Δhinge format presented in this study.
Figures
Similar articles
-
Treatment of myasthenia gravis by preventing acetylcholine receptor modulation.Ann N Y Acad Sci. 2008;1132:174-9. doi: 10.1196/annals.1405.034. Ann N Y Acad Sci. 2008. PMID: 18567867
-
Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice.Brain. 2012 Apr;135(Pt 4):1081-101. doi: 10.1093/brain/aws025. Epub 2012 Mar 6. Brain. 2012. PMID: 22396395
-
Pathogenic immune mechanisms at the neuromuscular synapse: the role of specific antibody-binding epitopes in myasthenia gravis.J Intern Med. 2014 Jan;275(1):12-26. doi: 10.1111/joim.12163. Epub 2013 Nov 29. J Intern Med. 2014. PMID: 24215230 Review.
-
How the autoimmune response to acetylcholine receptor impairs neuromuscular transmission in myasthenia gravis and its animal model.Fed Proc. 1978 Dec;37(14):2828-30. Fed Proc. 1978. PMID: 214352 No abstract available.
-
Pathogenic IgG4 subclass autoantibodies in MuSK myasthenia gravis.Ann N Y Acad Sci. 2012 Dec;1275:114-22. doi: 10.1111/j.1749-6632.2012.06808.x. Ann N Y Acad Sci. 2012. PMID: 23278586 Review.
Cited by
-
Characterization of an anti-fetal AChR monoclonal antibody isolated from a myasthenia gravis patient.Sci Rep. 2017 Oct 31;7(1):14426. doi: 10.1038/s41598-017-14350-8. Sci Rep. 2017. PMID: 29089519 Free PMC article.
-
Receptor clustering and pathogenic complement activation in myasthenia gravis depend on synergy between antibodies with multiple subunit specificities.Acta Neuropathol. 2022 Nov;144(5):1005-1025. doi: 10.1007/s00401-022-02493-6. Epub 2022 Sep 8. Acta Neuropathol. 2022. PMID: 36074148 Free PMC article.
-
New, Old, and Shared Antibody Specificities in Autoimmune Diseases.Antibodies (Basel). 2024 Mar 13;13(1):23. doi: 10.3390/antib13010023. Antibodies (Basel). 2024. PMID: 38534212 Free PMC article.
-
Myasthenia Gravis: Pathogenic Effects of Autoantibodies on Neuromuscular Architecture.Cells. 2019 Jul 2;8(7):671. doi: 10.3390/cells8070671. Cells. 2019. PMID: 31269763 Free PMC article. Review.
-
Taking the Hinge off: An Approach to Effector-Less Monoclonal Antibodies.Antibodies (Basel). 2020 Sep 23;9(4):50. doi: 10.3390/antib9040050. Antibodies (Basel). 2020. PMID: 32977708 Free PMC article.
References
-
- Mitra AK, McCarthy MP, Stroud RM. Three-dimensional structure of the nicotinic acetylcholine receptor and location of the major associated 43-kD cytoskeletal protein, determined at 22 A by low dose electron microscopy and x-ray diffraction to 12.5 A. J Cell Biol. 1989;109:755–774. doi: 10.1083/jcb.109.2.755. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
