

Physiological Functions of the Cellular Prion Protein
- PMID: 28428956
- PMCID: PMC5382174
- DOI: 10.3389/fmolb.2017.00019
Physiological Functions of the Cellular Prion Protein
Abstract
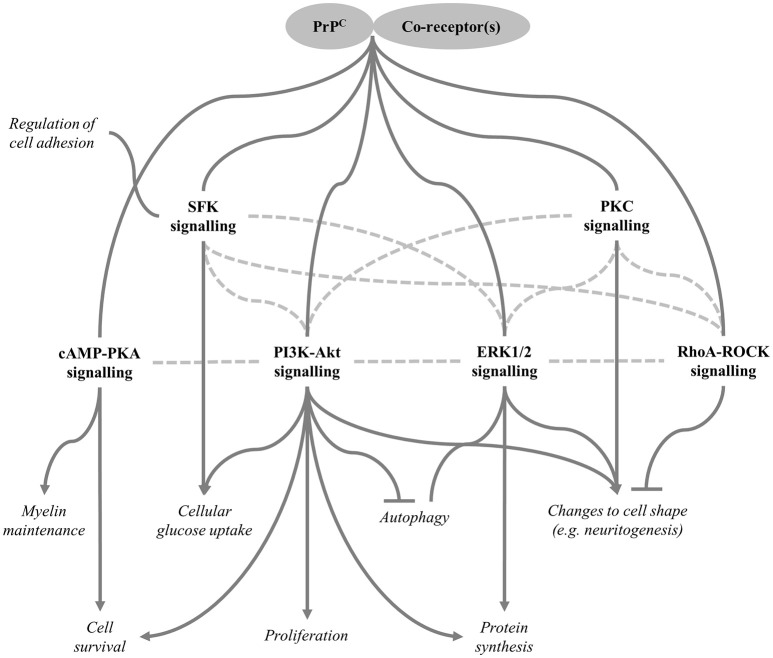
The prion protein, PrPC, is a small, cell-surface glycoprotein notable primarily for its critical role in pathogenesis of the neurodegenerative disorders known as prion diseases. A hallmark of prion diseases is the conversion of PrPC into an abnormally folded isoform, which provides a template for further pathogenic conversion of PrPC, allowing disease to spread from cell to cell and, in some circumstances, to transfer to a new host. In addition to the putative neurotoxicity caused by the misfolded form(s), loss of normal PrPC function could be an integral part of the neurodegenerative processes and, consequently, significant research efforts have been directed toward determining the physiological functions of PrPC. In this review, we first summarise important aspects of the biochemistry of PrPC before moving on to address the current understanding of the various proposed functions of the protein, including details of the underlying molecular mechanisms potentially involved in these functions. Over years of study, PrPC has been associated with a wide array of different cellular processes and many interacting partners have been suggested. However, recent studies have cast doubt on the previously well-established links between PrPC and processes such as stress-protection, copper homeostasis and neuronal excitability. Instead, the functions best-supported by the current literature include regulation of myelin maintenance and of processes linked to cellular differentiation, including proliferation, adhesion, and control of cell morphology. Intriguing connections have also been made between PrPC and the modulation of circadian rhythm, glucose homeostasis, immune function and cellular iron uptake, all of which warrant further investigation.
Keywords: PrPC; adhesion; differentiation; myelin maintenance; prion; proliferation; stress protection; transmissible spongiform encephalopathies.
Figures
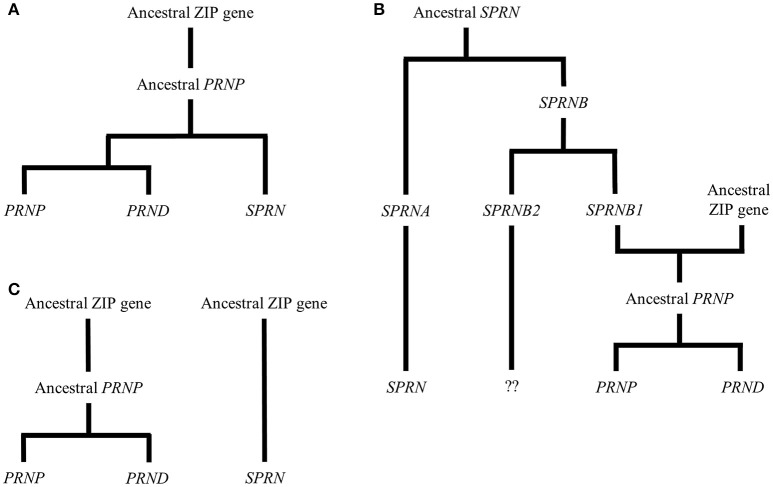
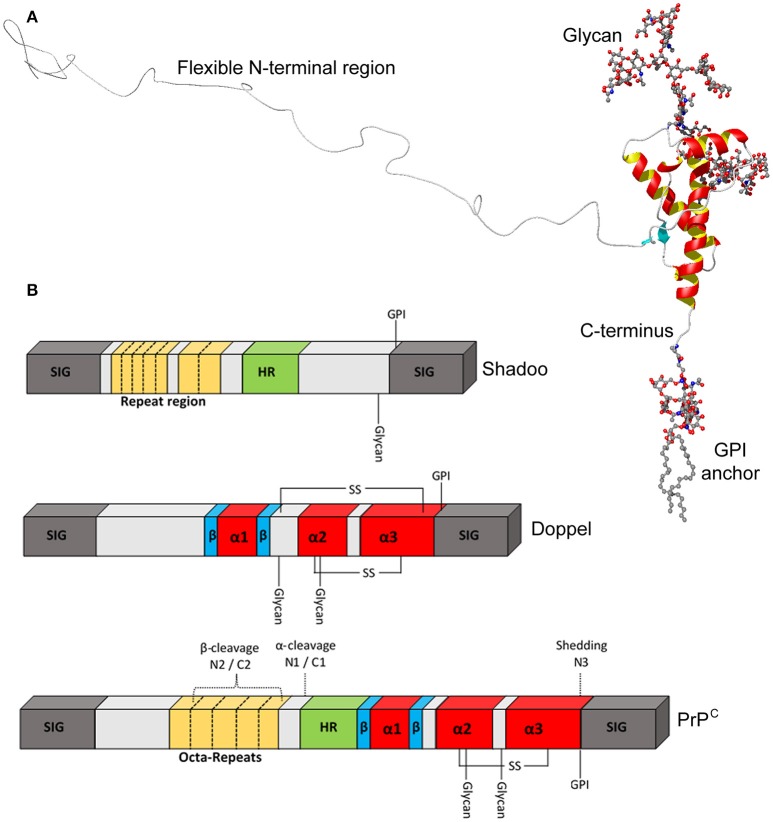
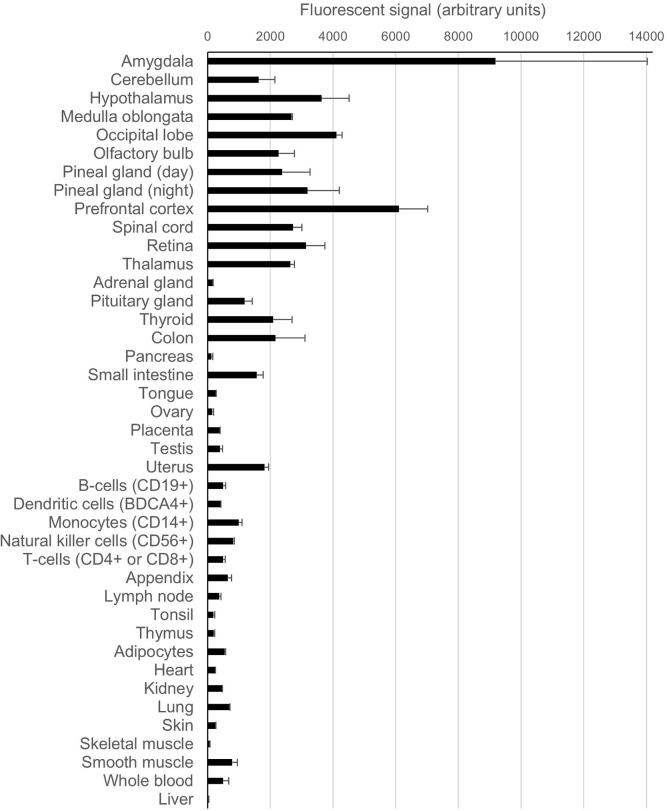
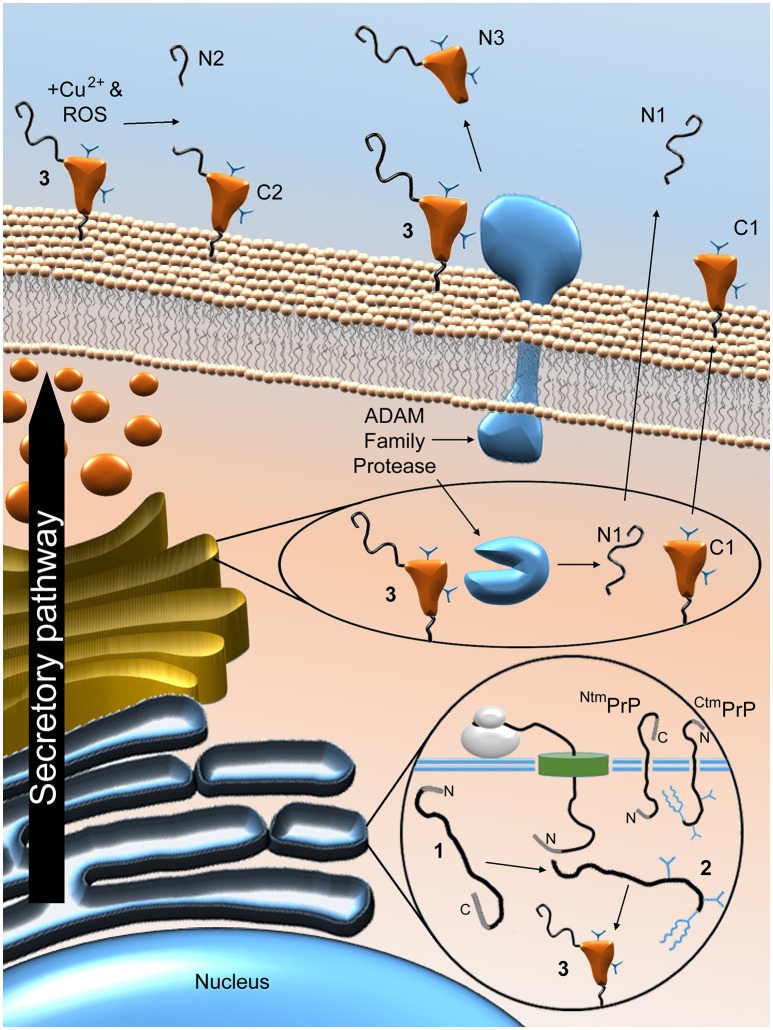
References
-
- Adle-Biassette H., Verney C., Peoc'h K., Dauge M. C., Razavi F., Choudat L., et al. (2006). Immunohistochemical expression of prion protein (PrPC) in the human forebrain during development. J. Neuropathol. Exp. Neurol. 65, 698–706. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous