

The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility
- PMID: 28429737
- PMCID: PMC5413954
- DOI: 10.1038/ncomms15074
The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility
Abstract
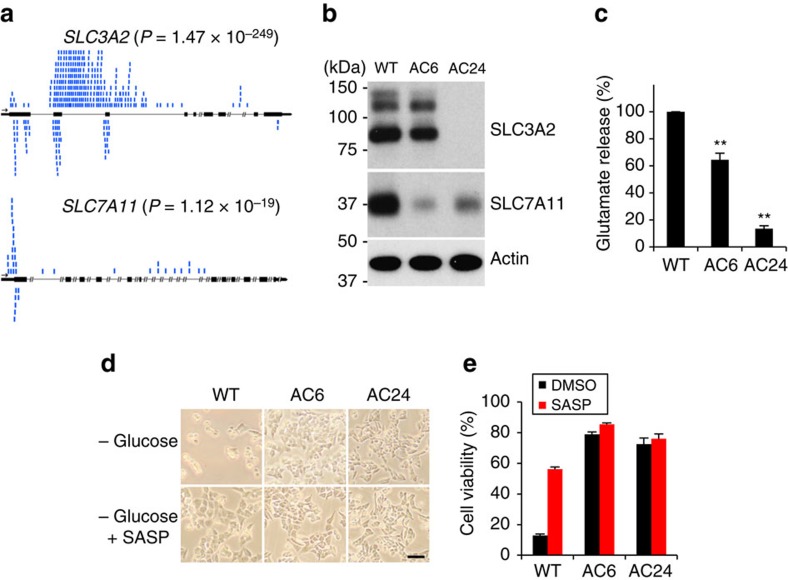
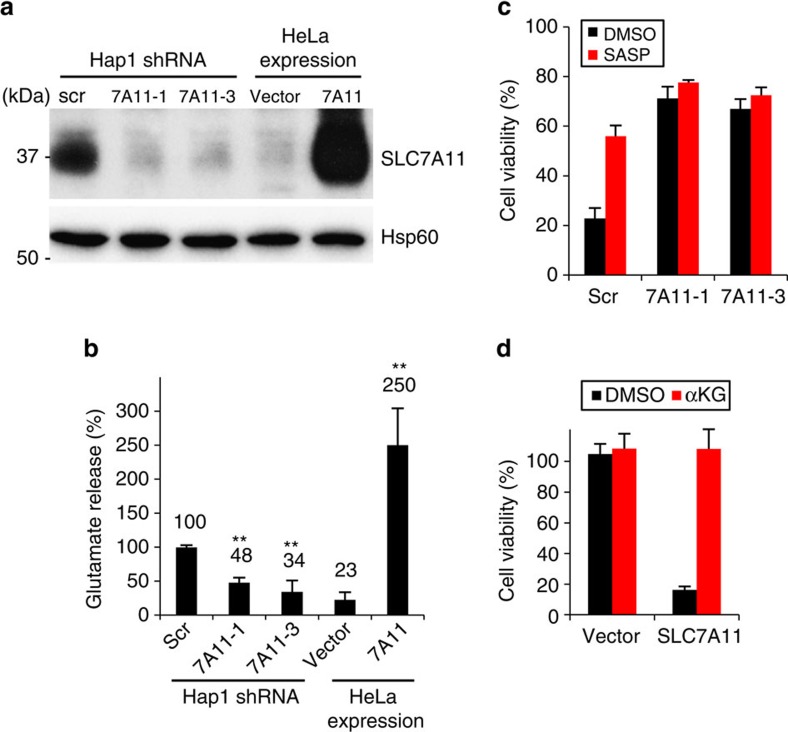
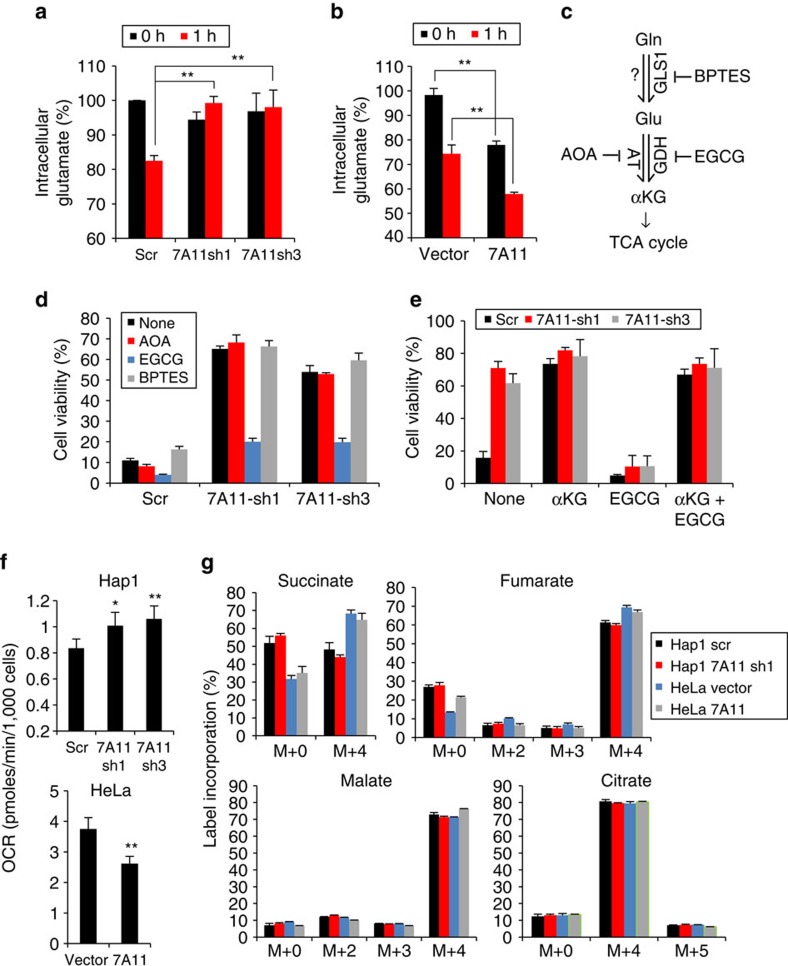
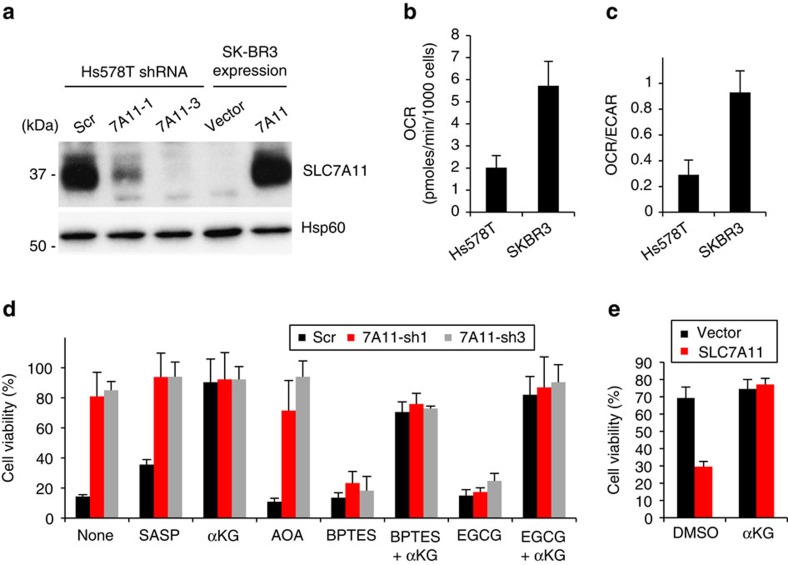
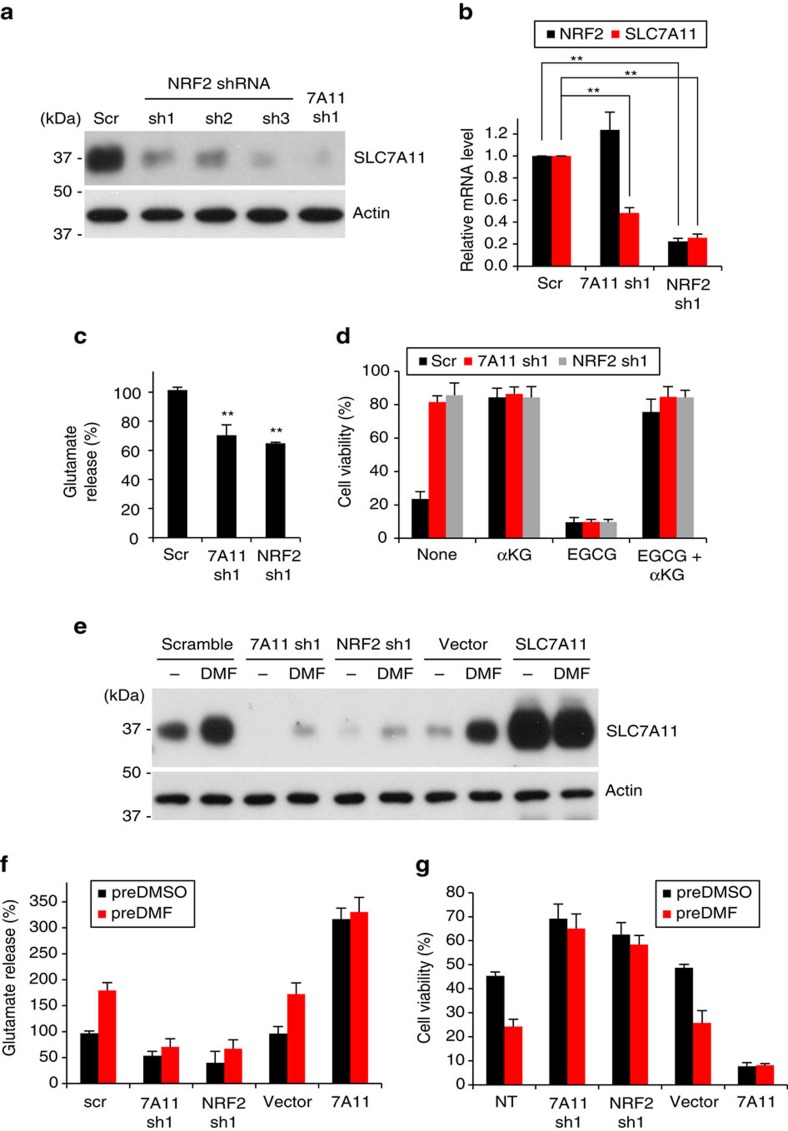
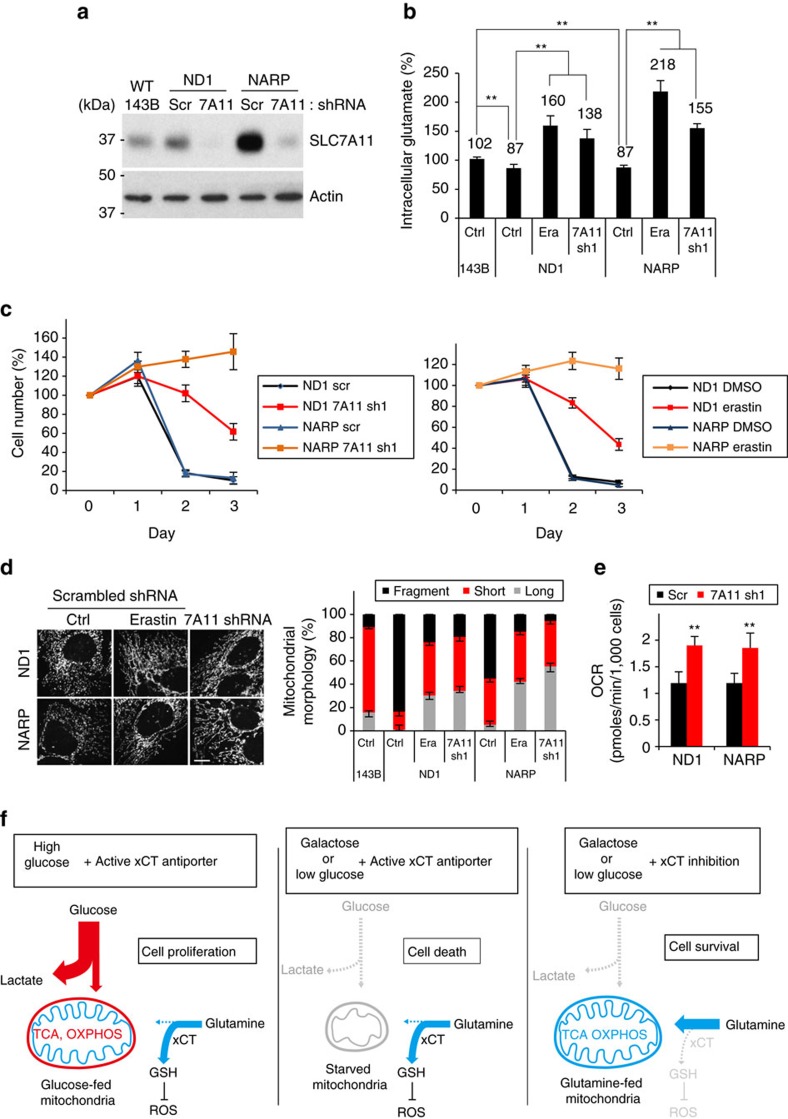
As noted by Warburg, many cancer cells depend on the consumption of glucose. We performed a genetic screen to identify factors responsible for glucose addiction and recovered the two subunits of the xCT antiporter (system xc-), which plays an antioxidant role by exporting glutamate for cystine. Disruption of the xCT antiporter greatly improves cell viability after glucose withdrawal, because conservation of glutamate enables cells to maintain mitochondrial respiration. In some breast cancer cells, xCT antiporter expression is upregulated through the antioxidant transcription factor Nrf2 and contributes to their requirement for glucose as a carbon source. In cells carrying patient-derived mitochondrial DNA mutations, the xCT antiporter is upregulated and its inhibition improves mitochondrial function and cell viability. Therefore, although upregulation of the xCT antiporter promotes antioxidant defence, it antagonizes glutamine metabolism and restricts nutrient flexibility. In cells with mitochondrial dysfunction, the potential utility of xCT antiporter inhibition should be further tested.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Levine A. J. & Puzio-Kuter A. M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330, 1340–1344 (2010). - PubMed
-
- DeBerardinis R. J., Lum J. J., Hatzivassiliou G. & Thompson C. B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
