

The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design
- PMID: 28430426
- PMCID: PMC5717763
- DOI: 10.1021/acs.jctc.7b00125
The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design
Erratum in
-
Correction to "The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design".J Chem Theory Comput. 2022 Jul 12;18(7):4594. doi: 10.1021/acs.jctc.2c00500. Epub 2022 Jun 6. J Chem Theory Comput. 2022. PMID: 35667008 No abstract available.
Abstract
Over the past decade, the Rosetta biomolecular modeling suite has informed diverse biological questions and engineering challenges ranging from interpretation of low-resolution structural data to design of nanomaterials, protein therapeutics, and vaccines. Central to Rosetta's success is the energy function: a model parametrized from small-molecule and X-ray crystal structure data used to approximate the energy associated with each biomolecule conformation. This paper describes the mathematical models and physical concepts that underlie the latest Rosetta energy function, called the Rosetta Energy Function 2015 (REF15). Applying these concepts, we explain how to use Rosetta energies to identify and analyze the features of biomolecular models. Finally, we discuss the latest advances in the energy function that extend its capabilities from soluble proteins to also include membrane proteins, peptides containing noncanonical amino acids, small molecules, carbohydrates, nucleic acids, and other macromolecules.
Figures
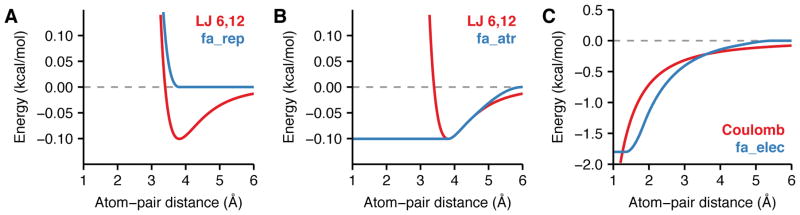
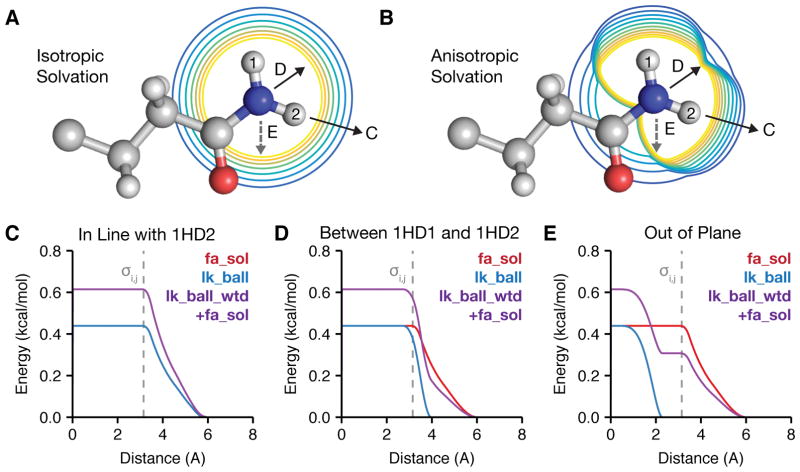
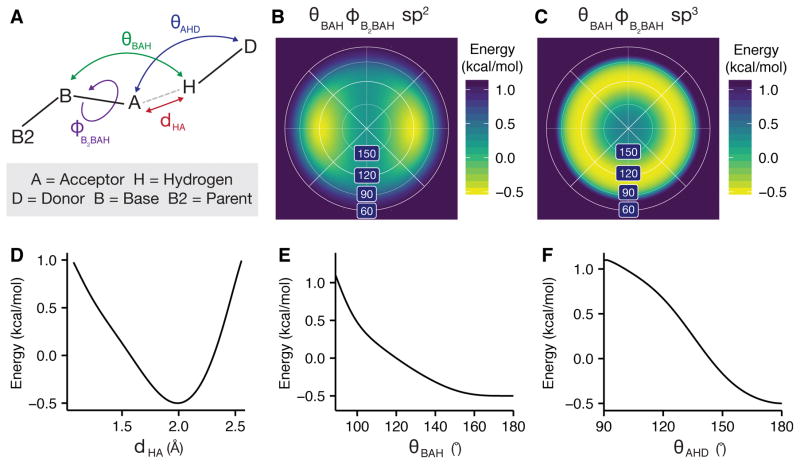
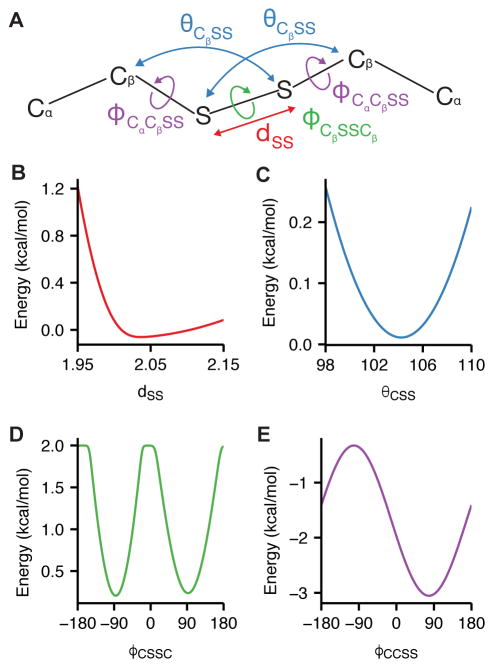
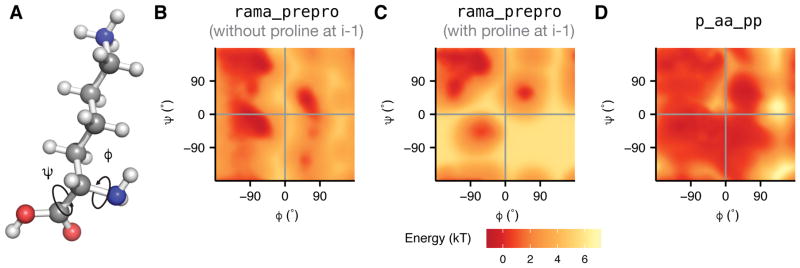
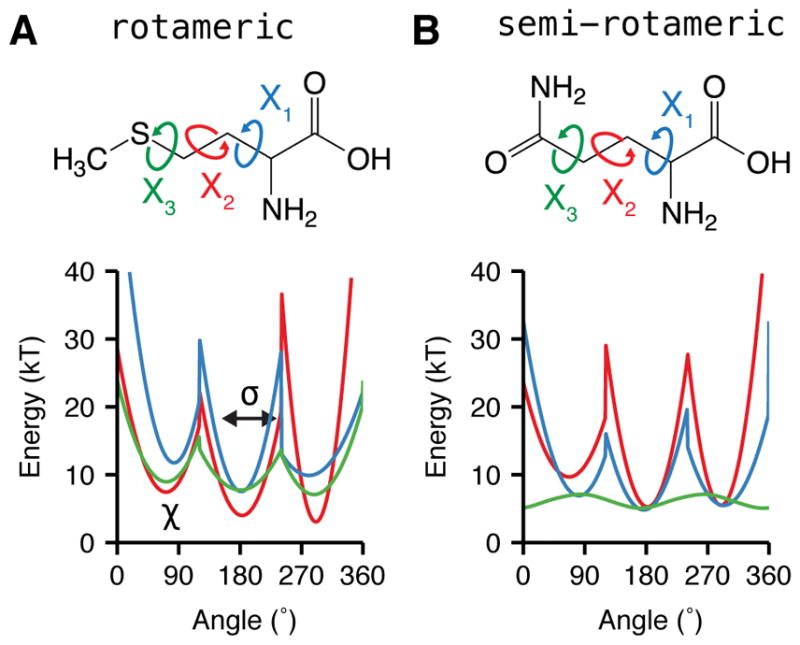
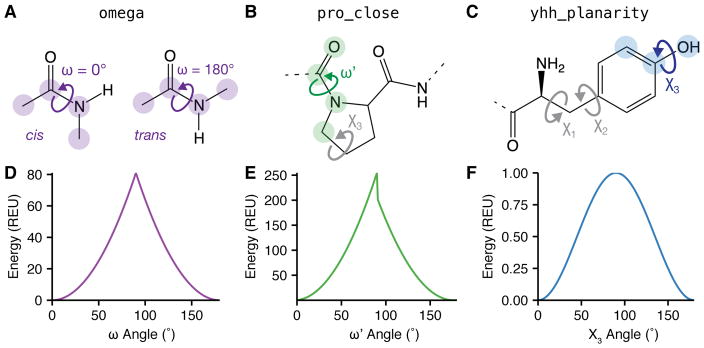
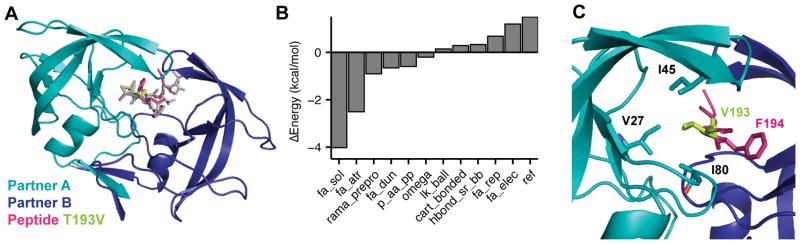
References
-
- Richardson JS. The Anatomy and Taxonomy of Protein Structure. Adv Protein Chem. 1981;34:167–339. - PubMed
-
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman KW, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban Y-EA, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods in enzymology. 2011;487:545–574. - PMC - PubMed
-
- Anfinsen CB. Principles That Govern the Folding of Protein Chains. Science. 1973;181(4096):223–230. - PubMed
-
- Lennard-Jones J. On the Determination of Molecular Fields I: From the Variation of Viscosity of a Gas with Temperature. R Soc London, Ser A, Contain Pap a Math Phys Character. 1924;106:441–462.
MeSH terms
Substances
Grants and funding
- R01 GM092802/GM/NIGMS NIH HHS/United States
- R01 GM073151/GM/NIGMS NIH HHS/United States
- K08 GM073141/GM/NIGMS NIH HHS/United States
- R01 GM078221/GM/NIGMS NIH HHS/United States
- F32 CA189246/CA/NCI NIH HHS/United States
- R01 GM110089/GM/NIGMS NIH HHS/United States
- R01 GM084453/GM/NIGMS NIH HHS/United States
- R35 GM122517/GM/NIGMS NIH HHS/United States
- F32 GM114961/GM/NIGMS NIH HHS/United States
- R01 GM073960/GM/NIGMS NIH HHS/United States
- R01 GM111819/GM/NIGMS NIH HHS/United States
- T32 GM008403/GM/NIGMS NIH HHS/United States
- R01 GM117189/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous