

doi: 10.1021/acs.jctc.6b01183.
Epub 2017 May 1.
Absolute Binding Free Energies between T4 Lysozyme and 141 Small Molecules: Calculations Based on Multiple Rigid Receptor Configurations
Affiliations
- PMID: 28430432
- PMCID: PMC5612505
- DOI: 10.1021/acs.jctc.6b01183
Item in Clipboard
Absolute Binding Free Energies between T4 Lysozyme and 141 Small Molecules: Calculations Based on Multiple Rigid Receptor Configurations
J Chem Theory Comput.
.
Abstract
We demonstrate the feasibility of estimating protein-ligand binding free energies using multiple rigid receptor configurations. On the basis of T4 lysozyme snapshots extracted from six alchemical binding free energy calculations with a flexible receptor, binding free energies were estimated for a total of 141 ligands. For 24 ligands, the calculations reproduced flexible-receptor estimates with a correlation coefficient of 0.90 and a root-mean-square error of 1.59 kcal/mol. The accuracy of calculations based on Poisson-Boltzmann/surface area implicit solvent was comparable to that of previously reported free energy calculations.
Figures
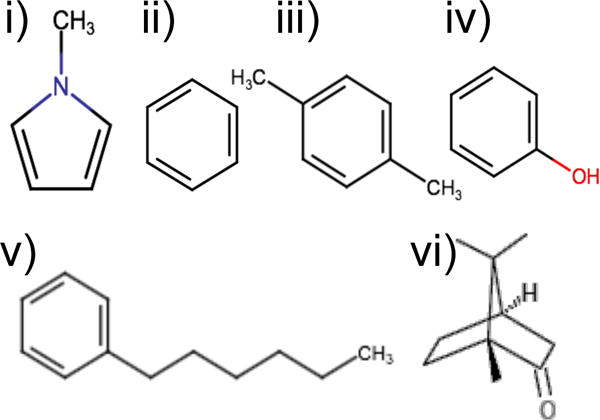
Ligands used in YANK calculations to generate snapshots for BPMF calculations: i) methylpyrrole, ii) benzene, iii) p-xylene, iv) phenol, v) n-hexylbenzene, and vi) (±)-camphor.
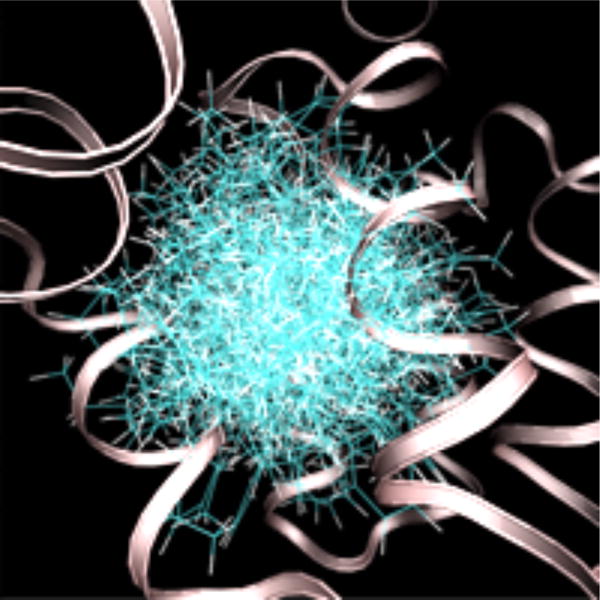
Configurations of n-hexylbenzene sampled at 600 K without a receptor interaction grid. The secondary structure of T4 lysozyme is shown as a ribbon. The protein structure is from an alchemical pathway calculation of DL-camphor binding.
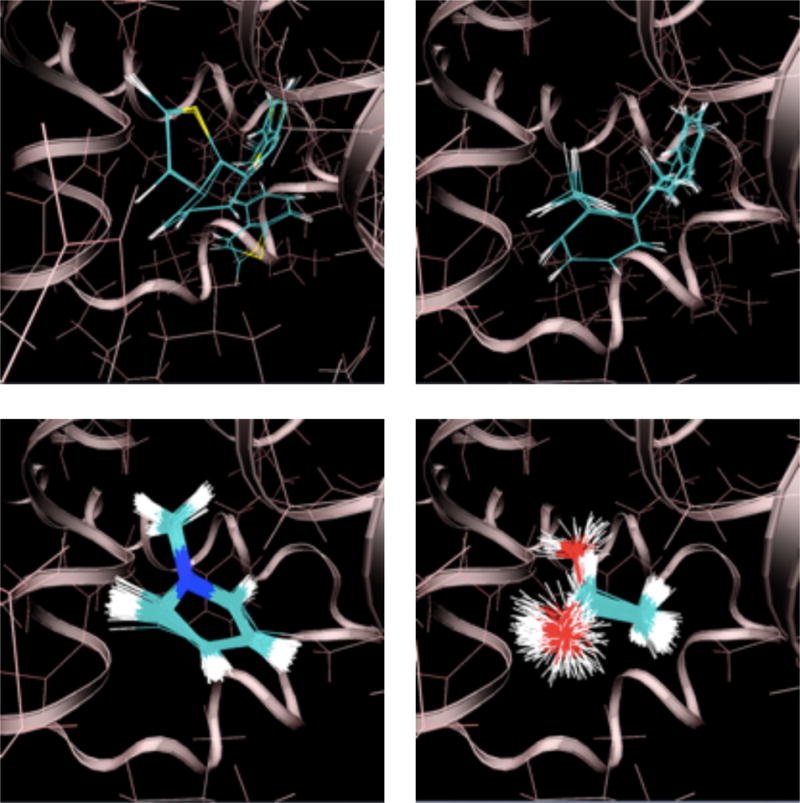
Ligand configurations sampled at 300 K with a full receptor interaction grid. Clockwise from the upper left: thianaphthene, o-xylene, 1-methylpyrrole, and ethanol. The secondary structure of T4 lysozyme is shown as a ribbon. The protein structure is from an alchemical pathway calculation of p-xylene binding.
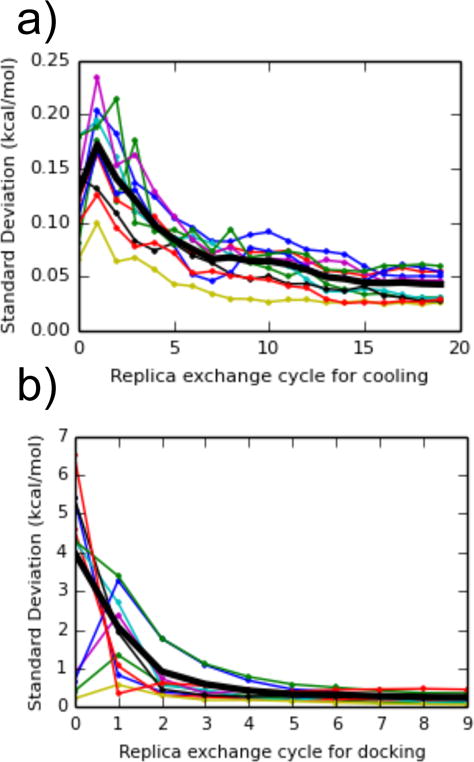
The standard deviation of free energy estimates as a function of the number of replica exchange cycles, for (a) changing the ligand temperature from 300 K to 600 K, and (b) scaling the receptor-ligand interaction grid from 0 to 1 while decreasing the temperature. Values were calculated between dipropyl disulfide, thianaphthene, isobutylbenzene, dibutyl-disulfide, phenylacetylene, cyclohexane, 1-heptanol, 1-propanol, 1,1-diethylurea, p-xylene, and a snapshot of T4 lysozyme from an alchemical pathway calculation with n-hexylbenzene. The thick black line is the average of the standard deviation across the 10 complexes.
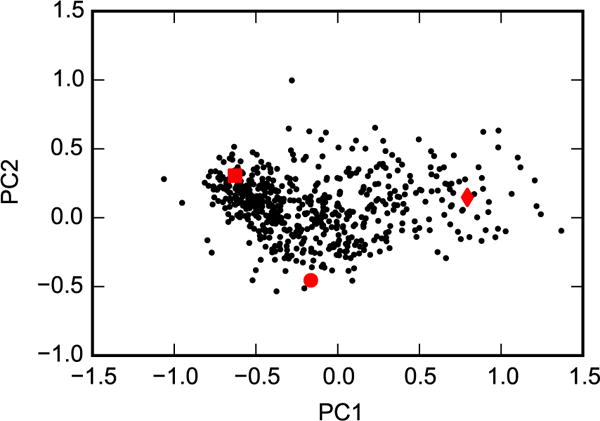
Principal components analysis was performed for the heavy atoms of helix F (residues 107 to 115) based on the crystal structures 2OTZ, 3DN3, and 1QUD, which represent closed (square), intermediate (circle), and open (diamond) conformations, respectively. The 576 representative snapshots (black dots) were then projected onto these eigenvectors.
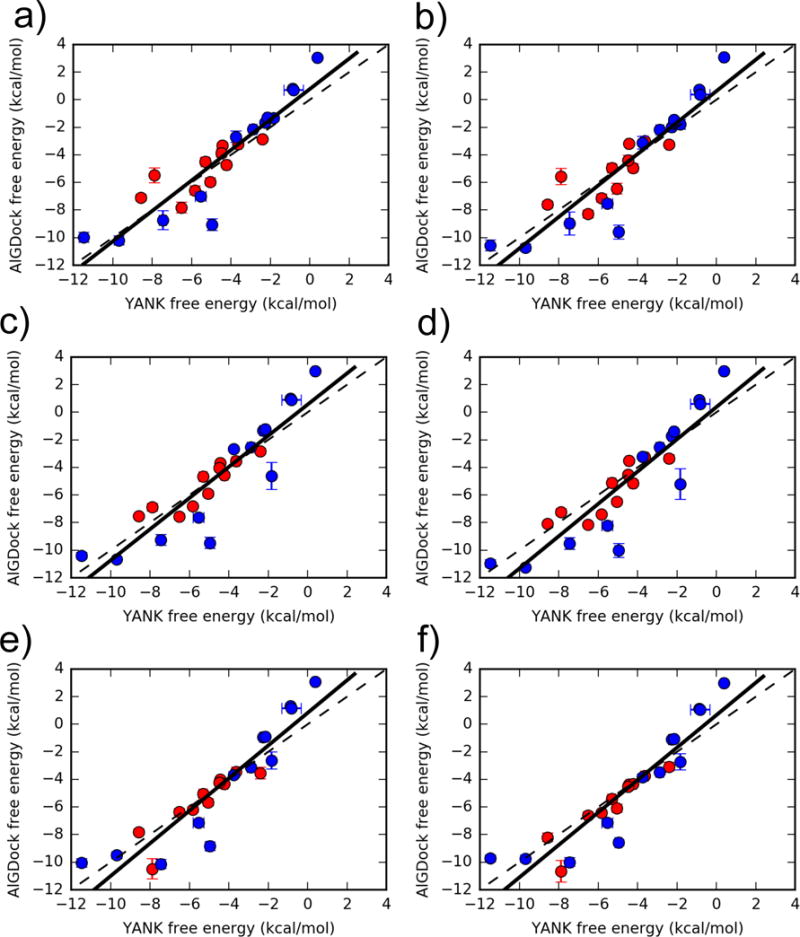
Binding free energies for 24 ligands estimated using YANK (x-axis) and AlGDock (y-axis) based on the OBC2 implicit solvent model. Active molecules are shown as red circles and inactive molecules as blue circles. The labels correspond to different weighting schemes (see text for details). Error bars denote the standard deviation from three independent YANK calculations (x-axis) or from bootstrapping BPMFs (y-axis), with the range of error bars representing a single standard deviation. The function y = x is shown as a dashed line and the linear regression for all ligands as a solid line.
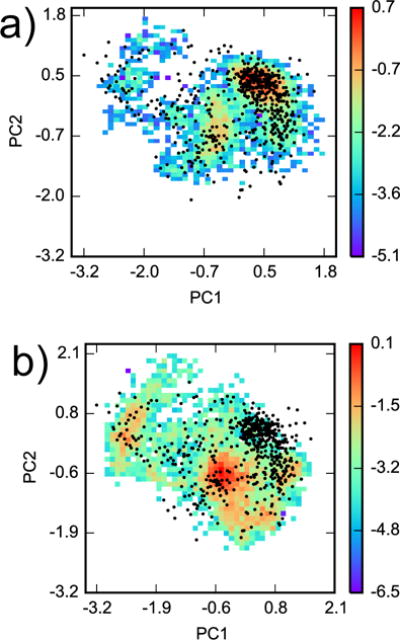
Configuration space comparison between YANK and AlGDock. Principal components analysis was performed on snapshots from all YANK simulations for heavy atoms within 5 Å of Val 111 in PDB ID 3DMZ. Two-dimensional histograms, weighted by Eq. 5, of YANK snapshots from (a) indole and (b) methanol were projected on the first two principal components. The histograms are plotted on a logarithmic scale. The black dots are projections of the 576 snapshots used in AlGDock calculations onto the same eigenvectors.
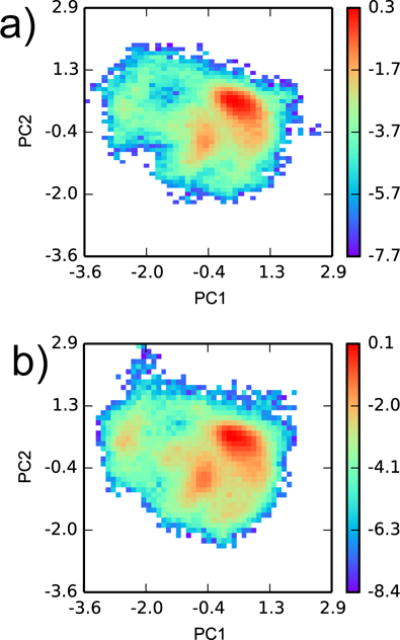
Configuration space comparison between YANK samples from (a) all active and (b) all inactive ligands. Principal components analysis was performed on snapshots from all YANK simulations for heavy atoms within 5 Å of Val 111 in PDB ID 3DMZ. Two-dimensional histograms, weighted by Eq. 5, of YANK snapshots were projected on the first two principal components. The histograms are plotted on a logarithmic scale.
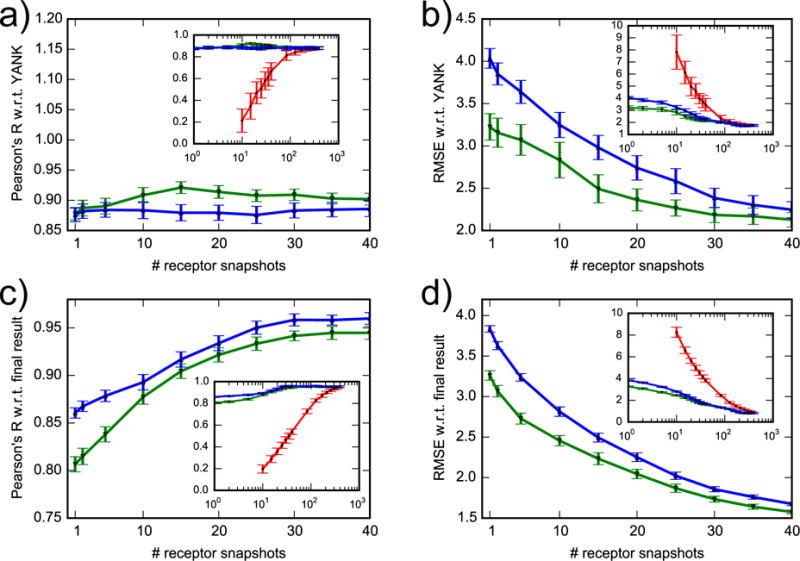
Correlation coefficient and RMSE of AlGDock free energy with respect to YANK (a, b) and to final result (c, d). Snapshots were selected randomly (red line), with lowest docking scores (green line) or with lowest BPMFs (blue line). The x-axis of the inset plots is on a log scale. For clarity, data for randomly selected snapshots are only shown for more than 10.
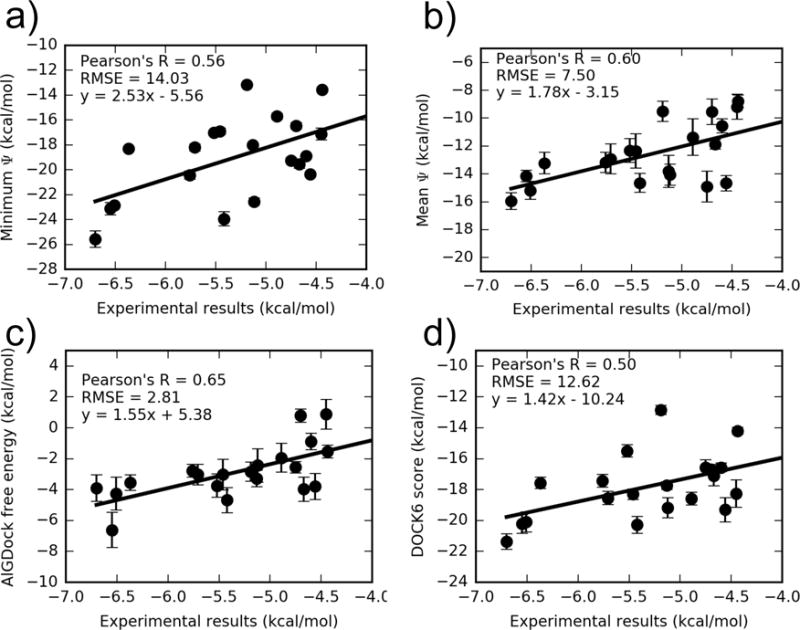
AlGDock free energy estimates in PBSA implicit solvent were based on BPMFs calculated with either the (a) minimum interaction energy, (b) mean interaction energy, or (c) MBAR estimator, using weighting scheme (c). A comparison of the average UCSF DOCK 6 grid score with experiment is shown in (d). Note that the y axes have different limits. The outlier iodobenzene is excluded. For the same plot with iodobenzene, see Fig. S5 in the SI.
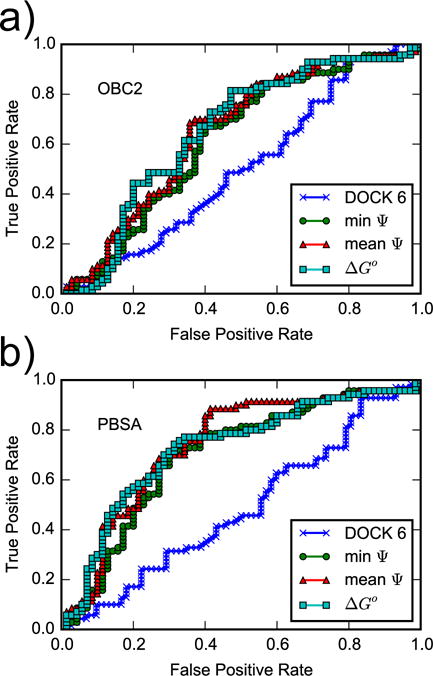
ROC curves for the DOCK 6 score and for AlGDock scores calculated using the (a) OBC2 or (b) PBSA implicit solvent models. Snapshots from YANK simulations with active ligands are weighted according to scheme (c).
Similar articles
-
Using the fast fourier transform in binding free energy calculations.J Comput Chem. 2018 Apr 30;39(11):621-636. doi: 10.1002/jcc.25139. Epub 2017 Dec 22. J Comput Chem. 2018. PMID: 29270990 Free PMC article.
-
Protein-ligand binding free energies from exhaustive docking.J Phys Chem B. 2012 Jun 14;116(23):6872-9. doi: 10.1021/jp212646s. Epub 2012 Apr 2. J Phys Chem B. 2012. PMID: 22432509
-
Nonbonded Force Field Parameters from MBIS Partitioning of the Molecular Electron Density Improve Binding Affinity Predictions of the T4-Lysozyme Double Mutant.J Chem Inf Model. 2024 Apr 22;64(8):3269-3277. doi: 10.1021/acs.jcim.3c01912. Epub 2024 Mar 28. J Chem Inf Model. 2024. PMID: 38546407
-
The tail lysozyme complex of bacteriophage T4.Int J Biochem Cell Biol. 2003 Jan;35(1):16-21. doi: 10.1016/s1357-2725(02)00098-5. Int J Biochem Cell Biol. 2003. PMID: 12467643 Review.
-
Structural and genetic analysis of the folding and function of T4 lysozyme.FASEB J. 1996 Jan;10(1):35-41. doi: 10.1096/fasebj.10.1.8566545. FASEB J. 1996. PMID: 8566545 Review.
Cited by
-
Implicit ligand theory for relative binding free energies: II. An estimator based on control variates.J Phys Commun. 2020 Nov;4(11):115010. doi: 10.1088/2399-6528/abcbac. Epub 2020 Nov 26. J Phys Commun. 2020. PMID: 33817346 Free PMC article.
-
On Restraints in End-Point Protein-Ligand Binding Free Energy Calculations.J Comput Chem. 2020 Mar 5;41(6):573-586. doi: 10.1002/jcc.26119. Epub 2019 Dec 10. J Comput Chem. 2020. PMID: 31821590 Free PMC article.
-
The SAMPL6 SAMPLing challenge: assessing the reliability and efficiency of binding free energy calculations.J Comput Aided Mol Des. 2020 May;34(5):601-633. doi: 10.1007/s10822-020-00290-5. Epub 2020 Jan 27. J Comput Aided Mol Des. 2020. PMID: 31984465 Free PMC article.
-
Alchemical Grid Dock (AlGDock): Binding Free Energy Calculations between Flexible Ligands and Rigid Receptors.J Comput Chem. 2020 Mar 15;41(7):715-730. doi: 10.1002/jcc.26036. Epub 2019 Aug 9. J Comput Chem. 2020. PMID: 31397498 Free PMC article.
-
Temperature artifacts in protein structures bias ligand-binding predictions.Chem Sci. 2021 Jul 13;12(34):11275-11293. doi: 10.1039/d1sc02751d. eCollection 2021 Sep 1. Chem Sci. 2021. PMID: 34667539 Free PMC article.
References
-
- Michel J, Essex JW. Prediction of Protein-Ligand Binding Affinity by Free Energy Simulations: Assumptions, Pitfalls and Expectations. J Comput-Aided Mol Des. 2010;24:639–658. - PubMed
-
- Gilson MK, Zhou H-X. Calculation of Protein-Ligand Binding Affinities. Annu Rev Biophys Biomol Struct. 2007;36:21–42. - PubMed
-
- Warren GL, Andrews CW, Capelli A-M, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A Critical Assessment of Docking Programs and Scoring Functions. J Med Chem. 2006;49:5912–5931. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources