

Molecular Basis for Exercise-Induced Fatigue: The Importance of Strictly Controlled Cellular Ca2+ Handling
- PMID: 28432118
- PMCID: PMC5793735
- DOI: 10.1101/cshperspect.a029710
Molecular Basis for Exercise-Induced Fatigue: The Importance of Strictly Controlled Cellular Ca2+ Handling
Abstract
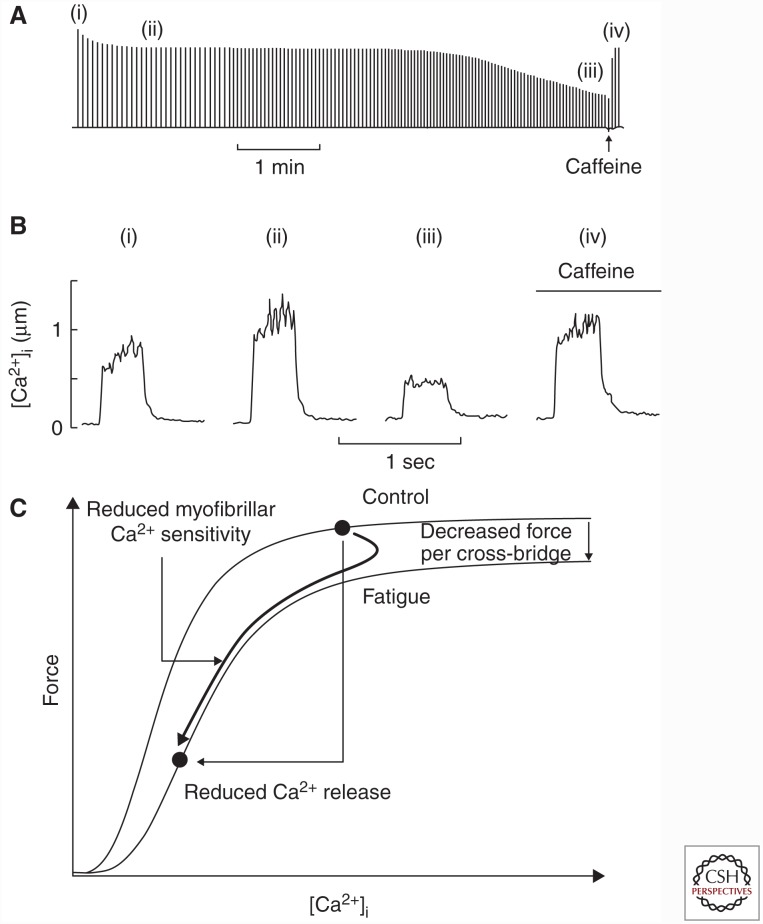
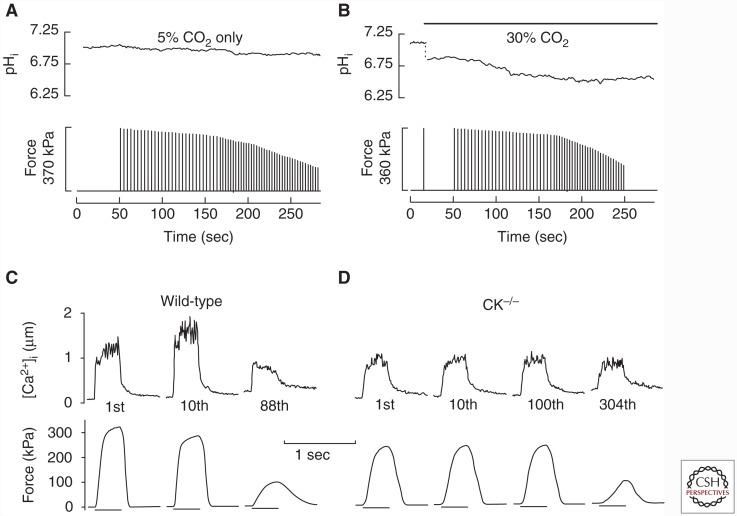
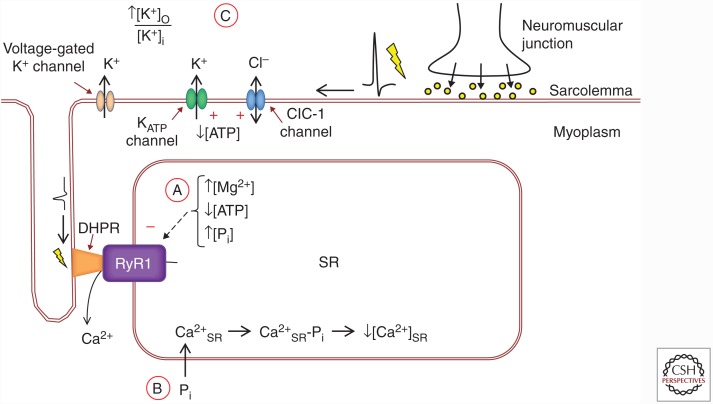
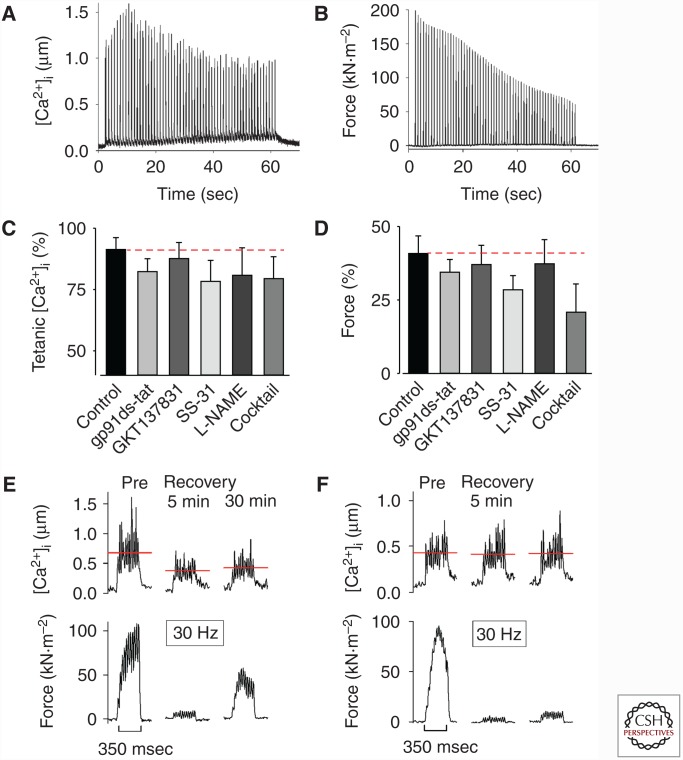
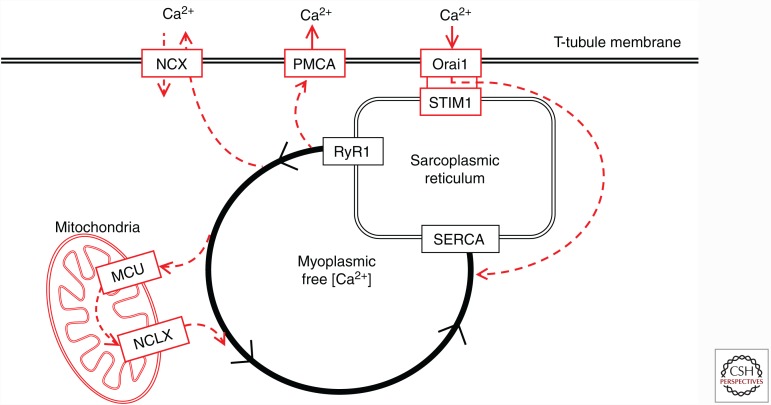
The contractile function of skeletal muscle declines during intense or prolonged physical exercise, that is, fatigue develops. Skeletal muscle fibers fatigue acutely during highly intense exercise when they have to rely on anaerobic metabolism. Early stages of fatigue involve impaired myofibrillar function, whereas decreased Ca2+ release from the sarcoplasmic reticulum (SR) becomes more important in later stages. SR Ca2+ release can also become reduced with more prolonged, lower intensity exercise, and it is then related to glycogen depletion. Increased reactive oxygen/nitrogen species can cause long-lasting impairments in SR Ca2+ release resulting in a prolonged force depression after exercise. In this article, we discuss molecular and cellular mechanisms of the above fatigue-induced changes, with special focus on multiple mechanisms to decrease SR Ca2+ release to avoid energy depletion and preserve muscle fiber integrity. We also discuss fatigue-related effects of exercise-induced Ca2+ fluxes over the sarcolemma and between the cytoplasm and mitochondria.
Copyright © 2018 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
Similar articles
-
Sarcoplasmic reticulum Ca(2+) release and muscle fatigue.J Appl Physiol (1985). 1999 Aug;87(2):471-83. doi: 10.1152/jappl.1999.87.2.471. J Appl Physiol (1985). 1999. PMID: 10444601 Review.
-
Reactive oxygen species and fatigue-induced prolonged low-frequency force depression in skeletal muscle fibres of rats, mice and SOD2 overexpressing mice.J Physiol. 2008 Jan 1;586(1):175-84. doi: 10.1113/jphysiol.2007.147470. Epub 2007 Nov 15. J Physiol. 2008. PMID: 18006575 Free PMC article.
-
Reactive oxygen/nitrogen species and contractile function in skeletal muscle during fatigue and recovery.J Physiol. 2016 Sep 15;594(18):5149-60. doi: 10.1113/JP270650. Epub 2016 Mar 20. J Physiol. 2016. PMID: 26857536 Free PMC article. Review.
-
A mitochondrial-targeted antioxidant improves myofilament Ca2+ sensitivity during prolonged low frequency force depression at low PO2.J Physiol. 2018 Mar 15;596(6):1079-1089. doi: 10.1113/JP275470. Epub 2018 Feb 11. J Physiol. 2018. PMID: 29334129 Free PMC article.
-
Prolonged exercise to fatigue in humans impairs skeletal muscle Na+-K+-ATPase activity, sarcoplasmic reticulum Ca2+ release, and Ca2+ uptake.J Appl Physiol (1985). 2004 Oct;97(4):1414-23. doi: 10.1152/japplphysiol.00964.2003. Epub 2004 May 21. J Appl Physiol (1985). 2004. PMID: 15155714 Clinical Trial.
Cited by
-
Calcium Fluxes in Work-Related Muscle Disorder: Implications from a Rat Model.Biomed Res Int. 2019 Sep 30;2019:5040818. doi: 10.1155/2019/5040818. eCollection 2019. Biomed Res Int. 2019. PMID: 31662979 Free PMC article.
-
Role of parvalbumin in fatigue-induced changes in force and cytosolic calcium transients in intact single mouse myofibers.J Appl Physiol (1985). 2022 Apr 1;132(4):1041-1053. doi: 10.1152/japplphysiol.00861.2021. Epub 2022 Mar 3. J Appl Physiol (1985). 2022. PMID: 35238653 Free PMC article.
-
Muscle Contractile Characteristics During Exhaustive Dynamic Exercise and Recovery.Front Physiol. 2021 Jul 2;12:660099. doi: 10.3389/fphys.2021.660099. eCollection 2021. Front Physiol. 2021. PMID: 34276393 Free PMC article.
-
Immediate but not prolonged effects of submaximal eccentric vs concentric fatiguing protocols on the etiology of hamstrings' motor performance fatigue.Eur J Appl Physiol. 2024 Nov;124(11):3215-3226. doi: 10.1007/s00421-024-05466-7. Epub 2024 Jun 7. Eur J Appl Physiol. 2024. PMID: 38847870
-
Effects of reduced muscle glycogen on excitation-contraction coupling in rat fast-twitch muscle: a glycogen removal study.J Muscle Res Cell Motil. 2019 Dec;40(3-4):353-364. doi: 10.1007/s10974-019-09524-y. Epub 2019 Jun 24. J Muscle Res Cell Motil. 2019. PMID: 31236763
References
-
- Allen DG, Lamb GD, Westerblad H. 2008. Skeletal muscle fatigue: Cellular mechanisms. Physiol Rev 88: 287–332. - PubMed
-
- Allen DG, Clugston E, Petersen Y, Röder IV, Chapman B, Rudolf R. 2011. Interactions between intracellular calcium and phosphate in intact mouse muscle during fatigue. J Appl Physiol (1985) 111: 358–366. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous