

Deregulated BCL-2 family proteins impact on repair of DNA double-strand breaks and are targets to overcome radioresistance in lung cancer
- PMID: 28432456
- PMCID: PMC11819454
- DOI: 10.1007/s00432-017-2427-1
Deregulated BCL-2 family proteins impact on repair of DNA double-strand breaks and are targets to overcome radioresistance in lung cancer
Abstract
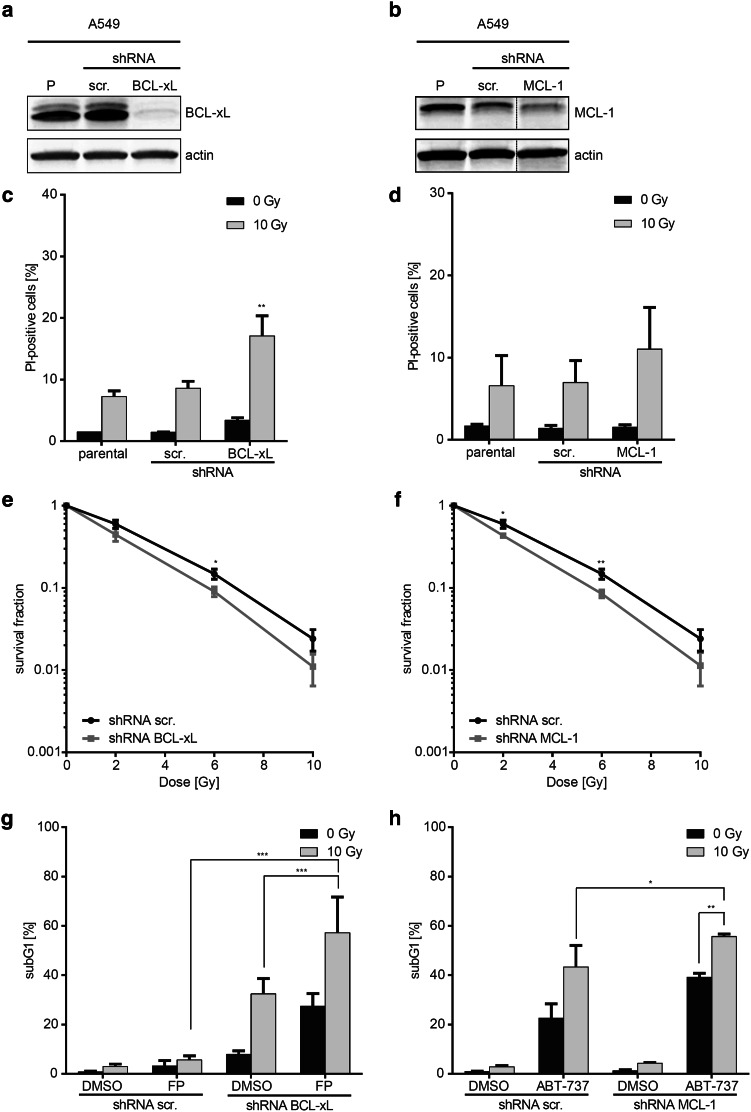
Purpose: DNA damage-induced cell death is a major effector mechanism of radiotherapy. Aberrant expression of anti-apoptotic BCL-2 family proteins is frequently observed in lung cancers. Against this background, we studied radioresistance mediated by BCL-2 family proteins at the mechanistic level and its potential as target for radiochemotherapy.
Methods: Lung cancer models stably expressing BCL-xL or MCL-1 were irradiated to study cell death, clonogenic survival, and DNA repair kinetics in vitro, and growth suppression of established tumors in vivo. Additionally, endogenous BCL-xL and MCL-1 were targeted by shRNA or pharmacologic agents prior to irradiation.
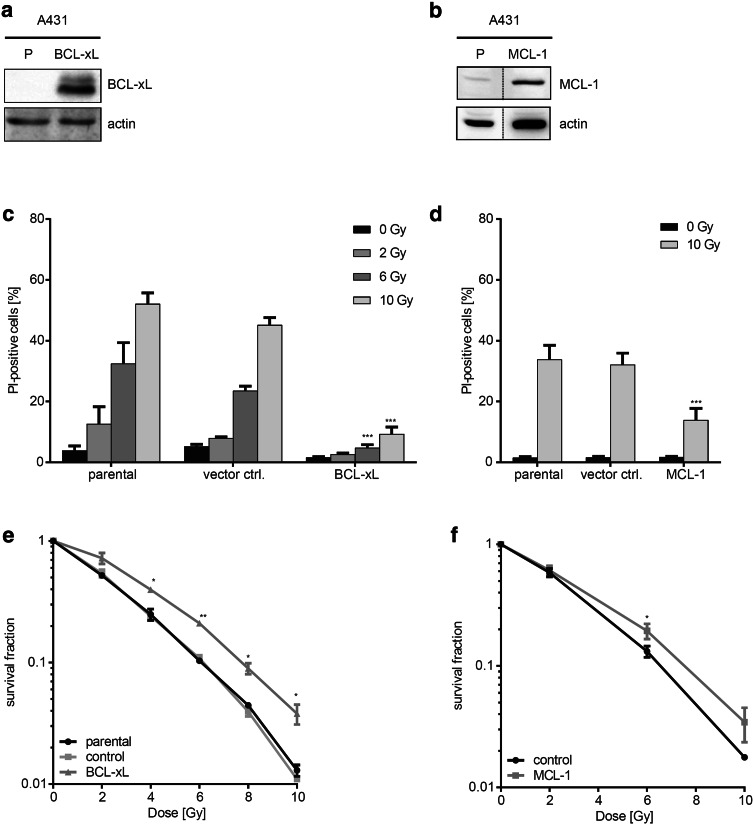
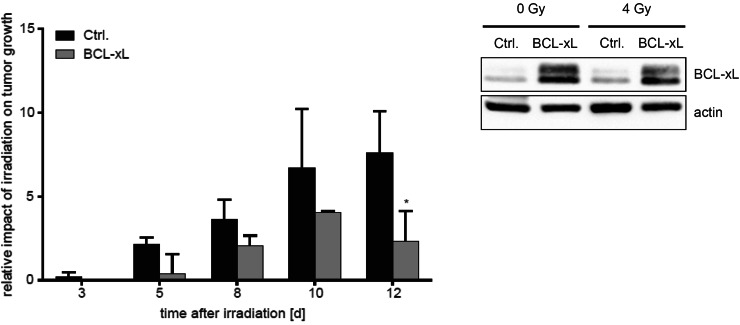
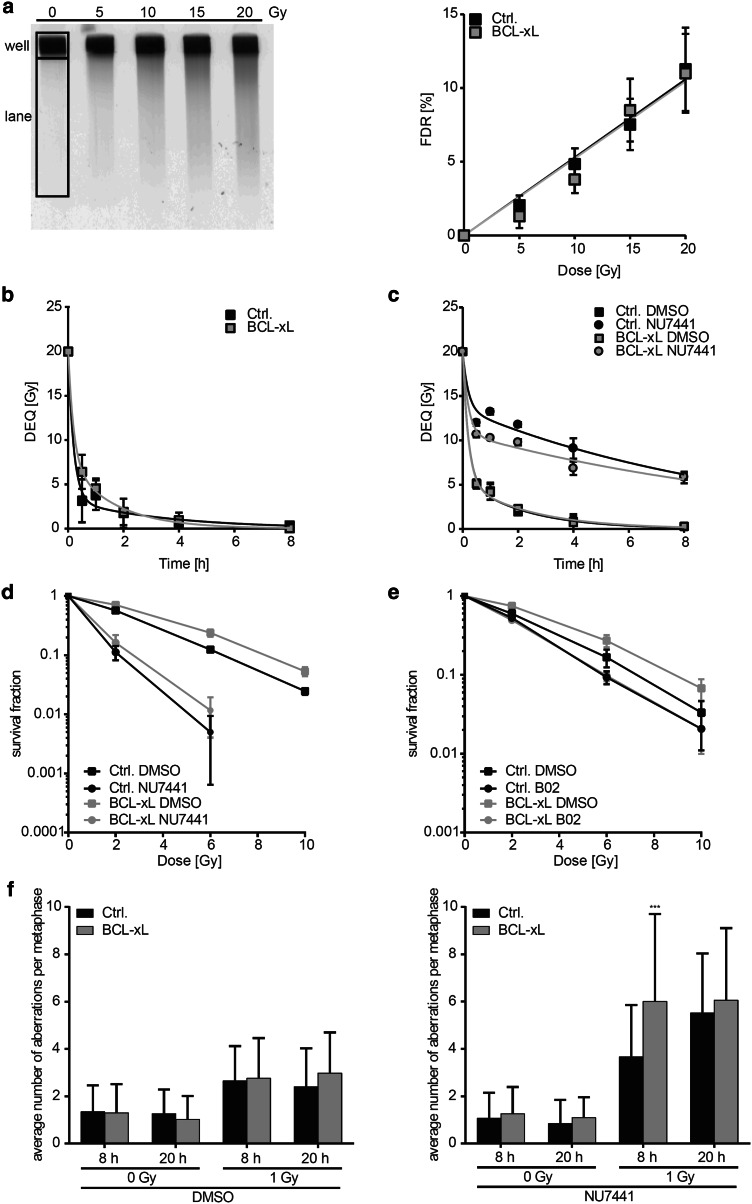
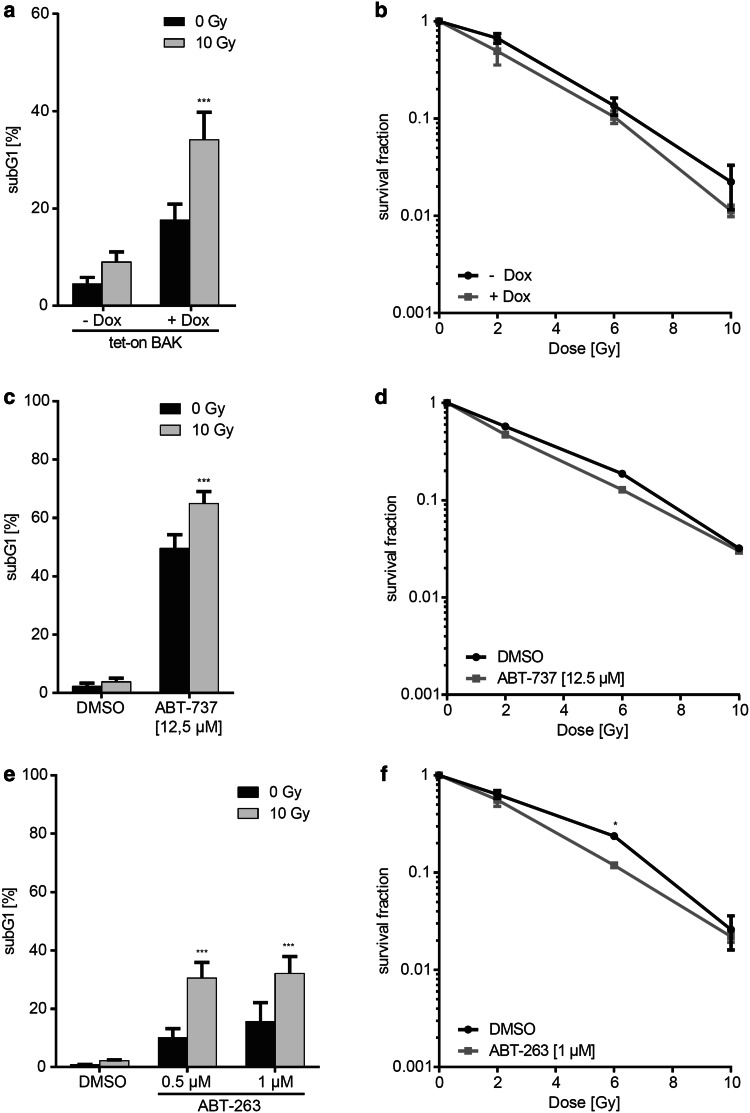
Results: Radiation exposure induced apoptosis at negligible levels. Yet, anti-apoptotic BCL-xL and MCL-1 expression conferred short-term and long-term radioresistance in vitro and in vivo. Radioresistance correlated with pertubations in homologous recombination repair and repair of DNA double-strand breaks by error-prone, alternative end-joining. Notably, genetic or pharmacologic targeting of BCL-xL or MCL-1 effectively sensitized lung cancer cells to radiotherapy.
Conclusions: In addition to directly suppressing apoptosis, BCL-2 family proteins confer long-term survival benefits to irradiated cancer cells associated with utilization of error-prone repair pathways. Targeting BCL-xL and MCL-1 is an attractive strategy for improving lung cancer radiotherapy.
Keywords: Alternative end-joining (alt-EJ); BCL-2 family; DNA double-strand break (DSB) repair; Homologous recombination repair (HRR); Lung cancer; Radioresistance.
Conflict of interest statement
The authors declare no conflicts of interest in relation with this work. The funding sources had no influence on the content of this manuscript.
Figures
Similar articles
-
Regulation of DNA repair mechanism in human glioma xenograft cells both in vitro and in vivo in nude mice.PLoS One. 2011;6(10):e26191. doi: 10.1371/journal.pone.0026191. Epub 2011 Oct 14. PLoS One. 2011. Retraction in: PLoS One. 2025 Jul 15;20(7):e0328089. doi: 10.1371/journal.pone.0328089. PMID: 22022560 Free PMC article. Retracted.
-
EGFR-mediated chromatin condensation protects KRAS-mutant cancer cells against ionizing radiation.Cancer Res. 2014 May 15;74(10):2825-34. doi: 10.1158/0008-5472.CAN-13-3157. Epub 2014 Mar 19. Cancer Res. 2014. PMID: 24648348 Free PMC article.
-
Artemis deficiency confers a DNA double-strand break repair defect and Artemis phosphorylation status is altered by DNA damage and cell cycle progression.DNA Repair (Amst). 2005 May 2;4(5):556-70. doi: 10.1016/j.dnarep.2005.02.001. DNA Repair (Amst). 2005. PMID: 15811628
-
A rapid and systematic review of the clinical effectiveness and cost-effectiveness of paclitaxel, docetaxel, gemcitabine and vinorelbine in non-small-cell lung cancer.Health Technol Assess. 2001;5(32):1-195. doi: 10.3310/hta5320. Health Technol Assess. 2001. PMID: 12065068
-
Role of Bcl-2 as a prognostic factor for survival in lung cancer: a systematic review of the literature with meta-analysis.Br J Cancer. 2003 Jul 7;89(1):55-64. doi: 10.1038/sj.bjc.6601095. Br J Cancer. 2003. PMID: 12838300 Free PMC article.
Cited by
-
Downregulation of CDC20 Increases Radiosensitivity through Mcl-1/p-Chk1-Mediated DNA Damage and Apoptosis in Tumor Cells.Int J Mol Sci. 2020 Sep 12;21(18):6692. doi: 10.3390/ijms21186692. Int J Mol Sci. 2020. PMID: 32932732 Free PMC article.
-
Targeting MCL-1 in cancer: current status and perspectives.J Hematol Oncol. 2021 Apr 21;14(1):67. doi: 10.1186/s13045-021-01079-1. J Hematol Oncol. 2021. PMID: 33883020 Free PMC article. Review.
-
TRAF4-mediated ubiquitination-dependent activation of JNK/Bcl-xL drives radioresistance.Cell Death Dis. 2023 Feb 10;14(2):102. doi: 10.1038/s41419-023-05637-y. Cell Death Dis. 2023. PMID: 36765039 Free PMC article.
-
Sonodynamic therapy induces oxidative stress, DNA damage and apoptosis in glioma cells.RSC Adv. 2018 Oct 25;8(63):36245-36256. doi: 10.1039/c8ra07099g. eCollection 2018 Oct 22. RSC Adv. 2018. PMID: 35558463 Free PMC article.
-
A novel role for apatinib in enhancing radiosensitivity in non-small cell lung cancer cells by suppressing the AKT and ERK pathways.PeerJ. 2021 Oct 28;9:e12356. doi: 10.7717/peerj.12356. eCollection 2021. PeerJ. 2021. PMID: 34760374 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
