

DNA single-strand break-induced DNA damage response causes heart failure
- PMID: 28436431
- PMCID: PMC5413978
- DOI: 10.1038/ncomms15104
DNA single-strand break-induced DNA damage response causes heart failure
Abstract
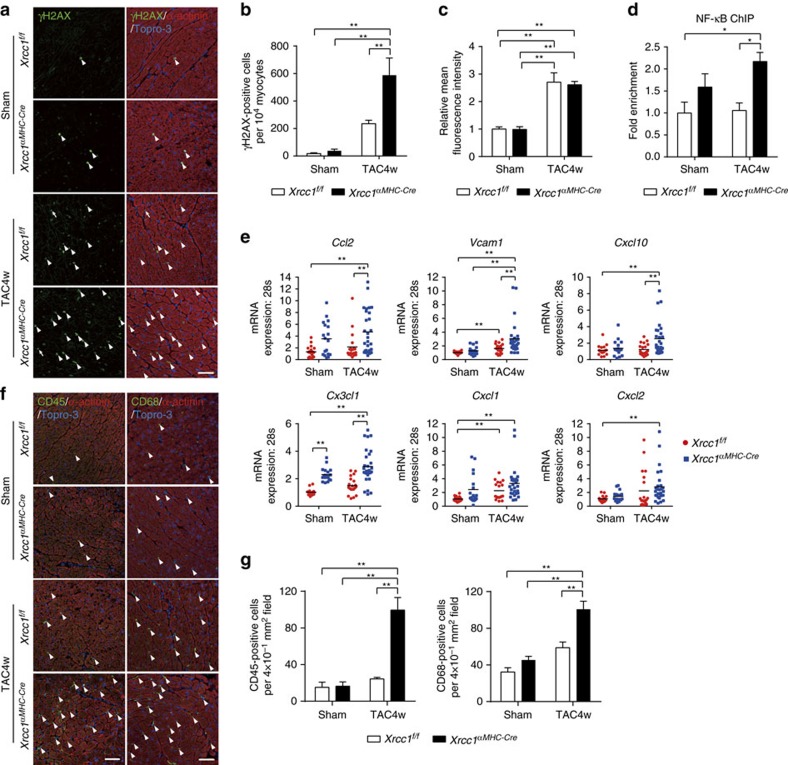
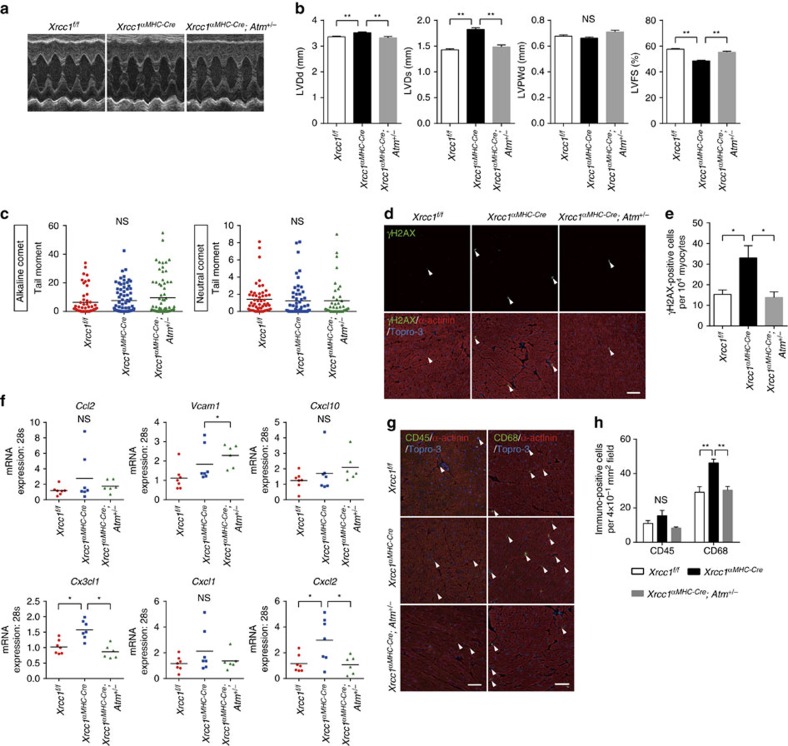
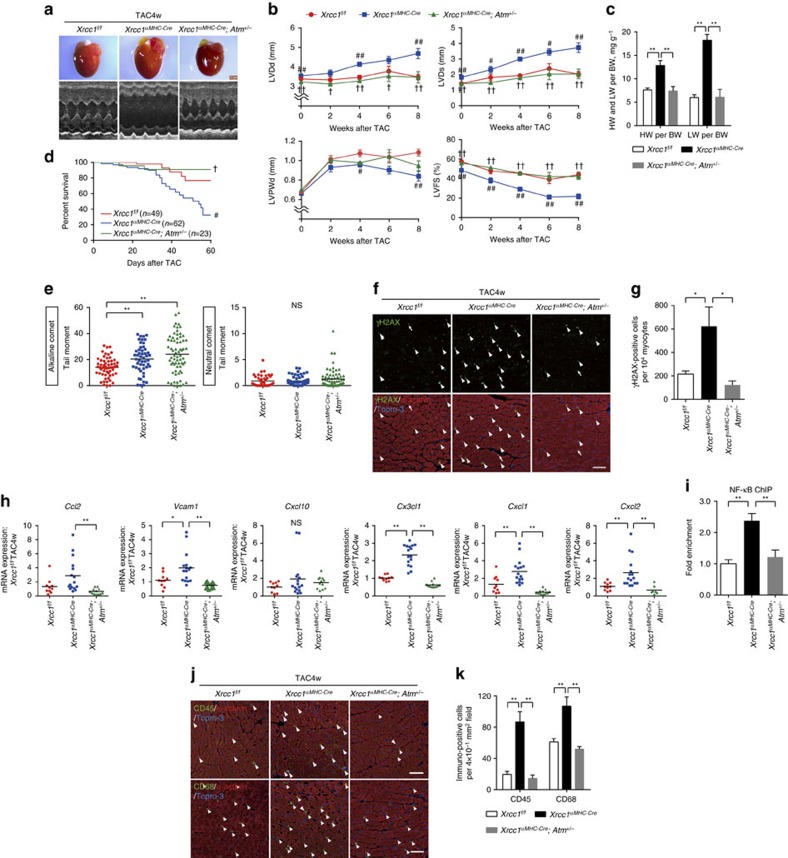
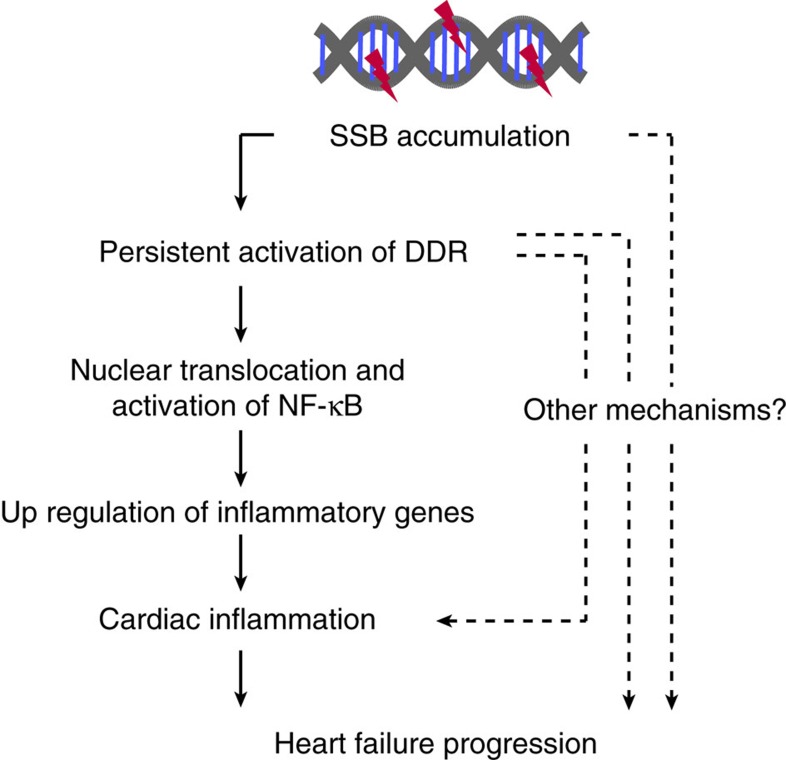
The DNA damage response (DDR) plays a pivotal role in maintaining genome integrity. DNA damage and DDR activation are observed in the failing heart, however, the type of DNA damage and its role in the pathogenesis of heart failure remain elusive. Here we show the critical role of DNA single-strand break (SSB) in the pathogenesis of pressure overload-induced heart failure. Accumulation of unrepaired SSB is observed in cardiomyocytes of the failing heart. Unrepaired SSB activates DDR and increases the expression of inflammatory cytokines through NF-κB signalling. Pressure overload-induced heart failure is more severe in the mice lacking XRCC1, an essential protein for SSB repair, which is rescued by blocking DDR activation through genetic deletion of ATM, suggesting the causative role of SSB accumulation and DDR activation in the pathogenesis of heart failure. Prevention of SSB accumulation or persistent DDR activation may become a new therapeutic strategy against heart failure.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Shiloh Y. & Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 (2013). - PubMed
-
- Sulli G., Di Micco R. & d'Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 12, 709–720 (2012). - PubMed
-
- Lee J. H. & Paull T. T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 26, 7741–7748 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
