

Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction
- PMID: 28436957
- PMCID: PMC5575773
- DOI: 10.1038/nm.4328
Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction
Abstract
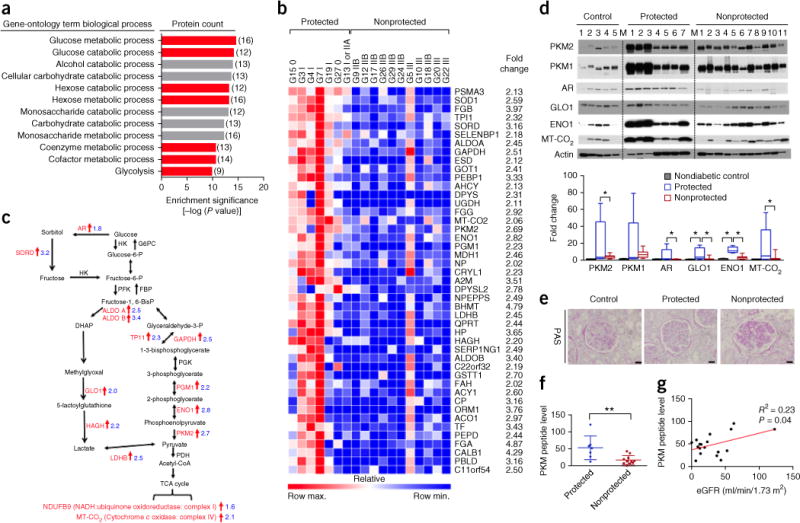
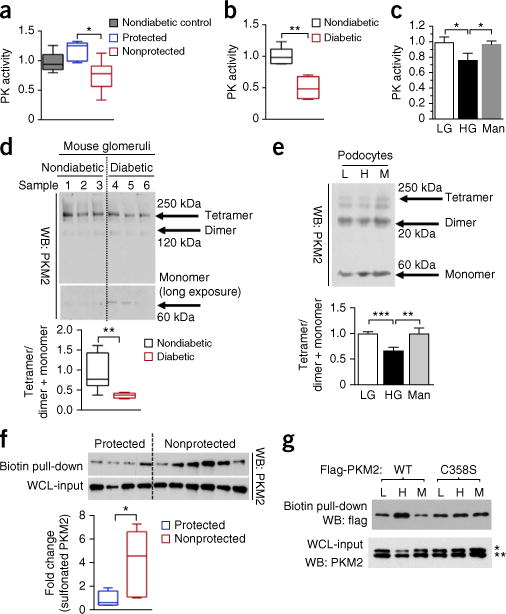
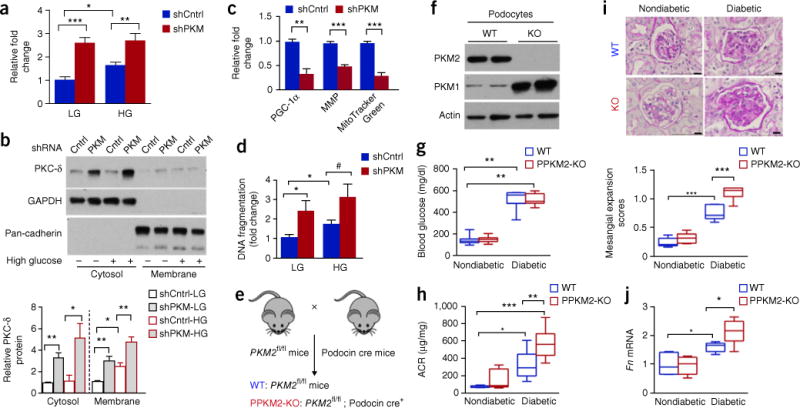
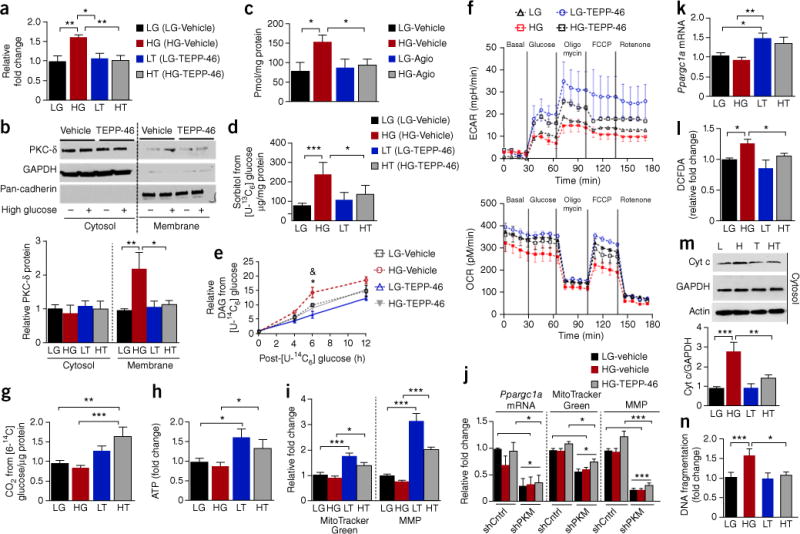
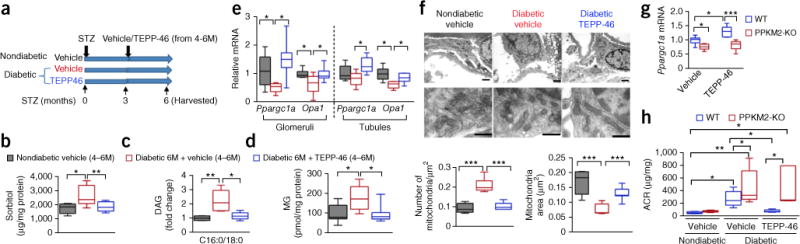
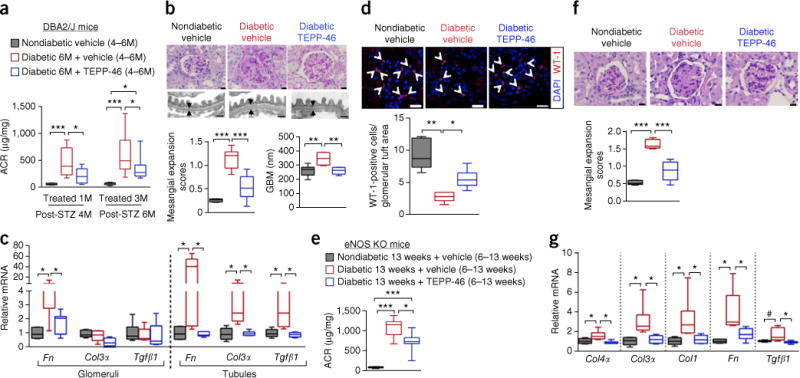
Diabetic nephropathy (DN) is a major cause of end-stage renal disease, and therapeutic options for preventing its progression are limited. To identify novel therapeutic strategies, we studied protective factors for DN using proteomics on glomeruli from individuals with extreme duration of diabetes (ł50 years) without DN and those with histologic signs of DN. Enzymes in the glycolytic, sorbitol, methylglyoxal and mitochondrial pathways were elevated in individuals without DN. In particular, pyruvate kinase M2 (PKM2) expression and activity were upregulated. Mechanistically, we showed that hyperglycemia and diabetes decreased PKM2 tetramer formation and activity by sulfenylation in mouse glomeruli and cultured podocytes. Pkm-knockdown immortalized mouse podocytes had higher levels of toxic glucose metabolites, mitochondrial dysfunction and apoptosis. Podocyte-specific Pkm2-knockout (KO) mice with diabetes developed worse albuminuria and glomerular pathology. Conversely, we found that pharmacological activation of PKM2 by a small-molecule PKM2 activator, TEPP-46, reversed hyperglycemia-induced elevation in toxic glucose metabolites and mitochondrial dysfunction, partially by increasing glycolytic flux and PGC-1α mRNA in cultured podocytes. In intervention studies using DBA2/J and Nos3 (eNos) KO mouse models of diabetes, TEPP-46 treatment reversed metabolic abnormalities, mitochondrial dysfunction and kidney pathology. Thus, PKM2 activation may protect against DN by increasing glucose metabolic flux, inhibiting the production of toxic glucose metabolites and inducing mitochondrial biogenesis to restore mitochondrial function.
Figures
Comment in
-
Diabetic nephropathy: Glucose metabolic flux in DN.Nat Rev Nephrol. 2017 Jul;13(7):384. doi: 10.1038/nrneph.2017.70. Epub 2017 May 15. Nat Rev Nephrol. 2017. PMID: 28502984 No abstract available.
-
Identification of a protective proteomic signature and a potential therapeutic target in diabetic nephropathy.Kidney Int. 2017 Oct;92(4):780-781. doi: 10.1016/j.kint.2017.08.002. Kidney Int. 2017. PMID: 28938946
References
-
- United States Renal Data System. 2015 USRDS Annual Data Report. USRDS; 2015.
-
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. - PubMed
-
- Nishikawa T, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
