

The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity
- PMID: 28438176
- PMCID: PMC5404317
- DOI: 10.1186/s13024-017-0174-z
The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity
Abstract
Background: Mutations in PINK1 and PARKIN are the most common causes of recessive early-onset Parkinson's disease (EOPD). Together, the mitochondrial ubiquitin (Ub) kinase PINK1 and the cytosolic E3 Ub ligase PARKIN direct a complex regulated, sequential mitochondrial quality control. Thereby, damaged mitochondria are identified and targeted to degradation in order to prevent their accumulation and eventually cell death. Homozygous or compound heterozygous loss of either gene function disrupts this protective pathway, though at different steps and by distinct mechanisms. While structure and function of PARKIN variants have been well studied, PINK1 mutations remain poorly characterized, in particular under endogenous conditions. A better understanding of the exact molecular pathogenic mechanisms underlying the pathogenicity is crucial for rational drug design in the future.
Methods: Here, we characterized the pathogenicity of the PINK1 p.I368N mutation on the clinical and genetic as well as on the structural and functional level in patients' fibroblasts and in cell-based, biochemical assays.
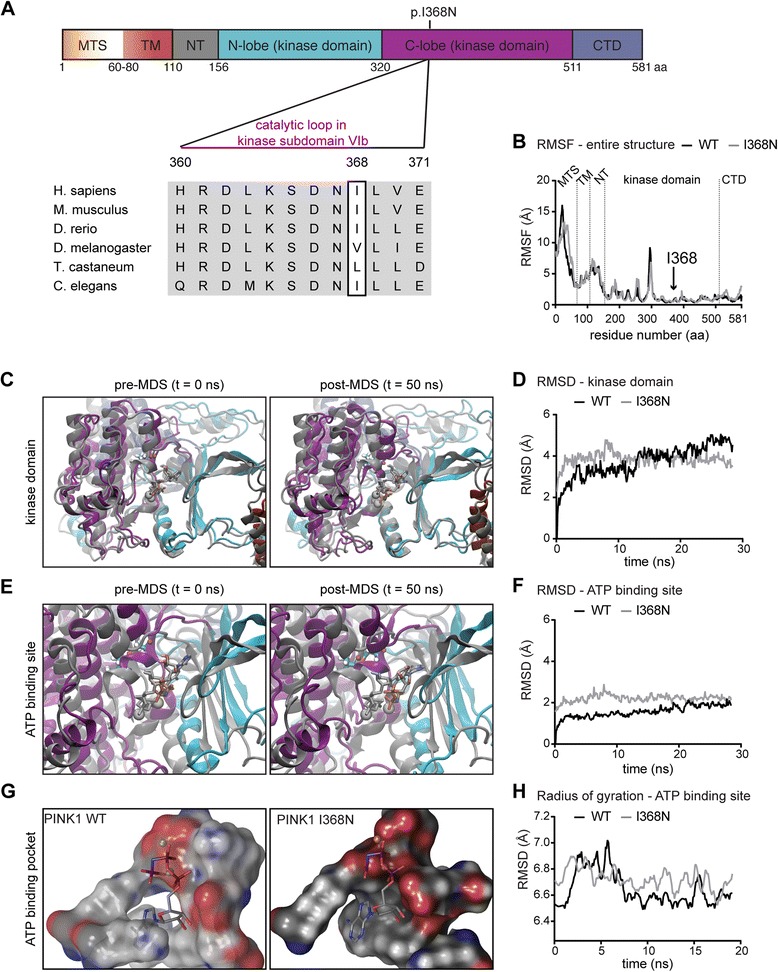
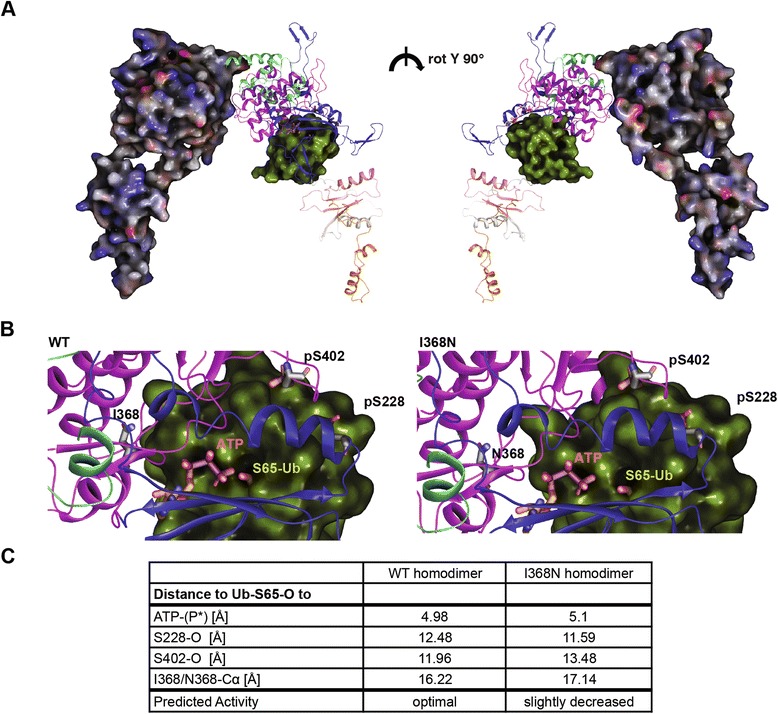
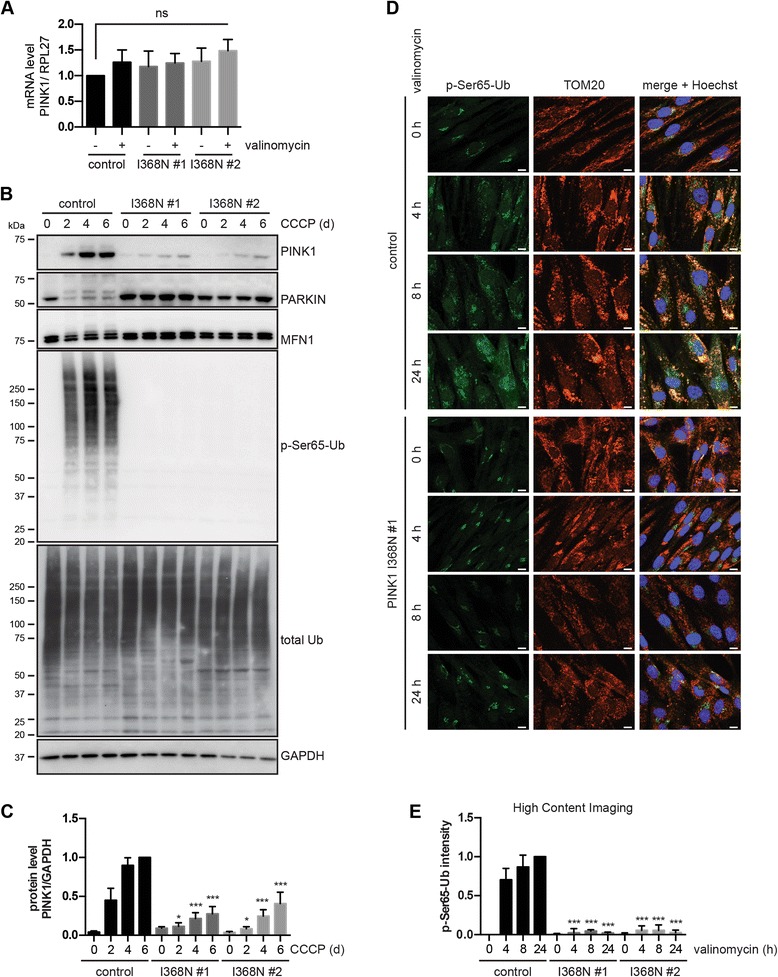
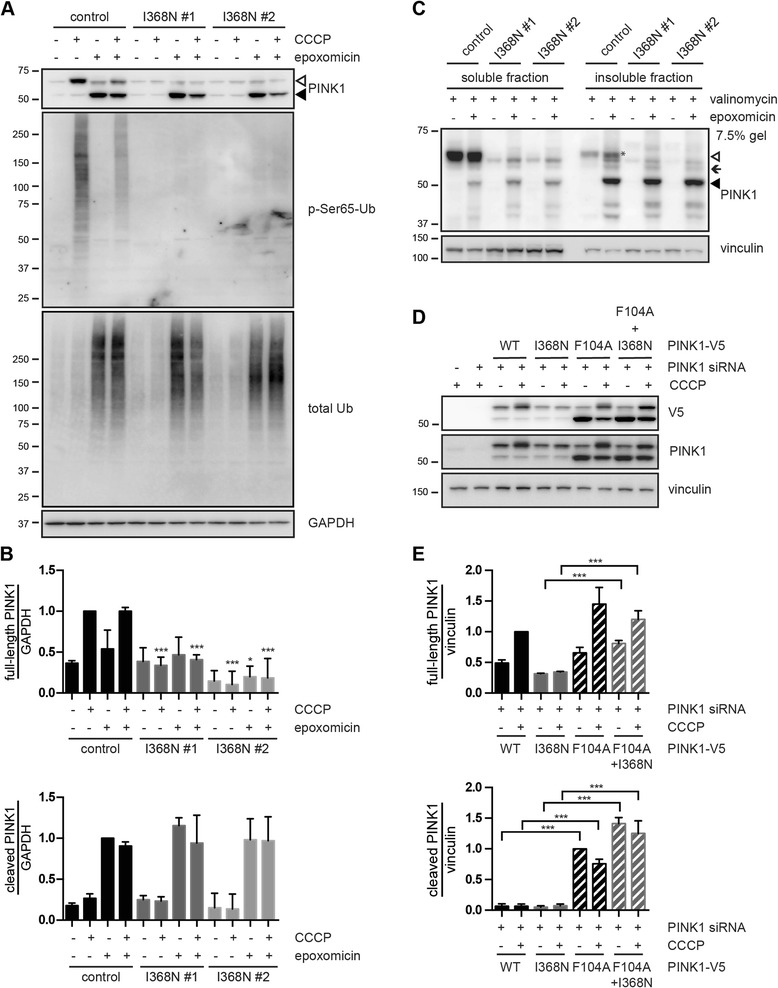
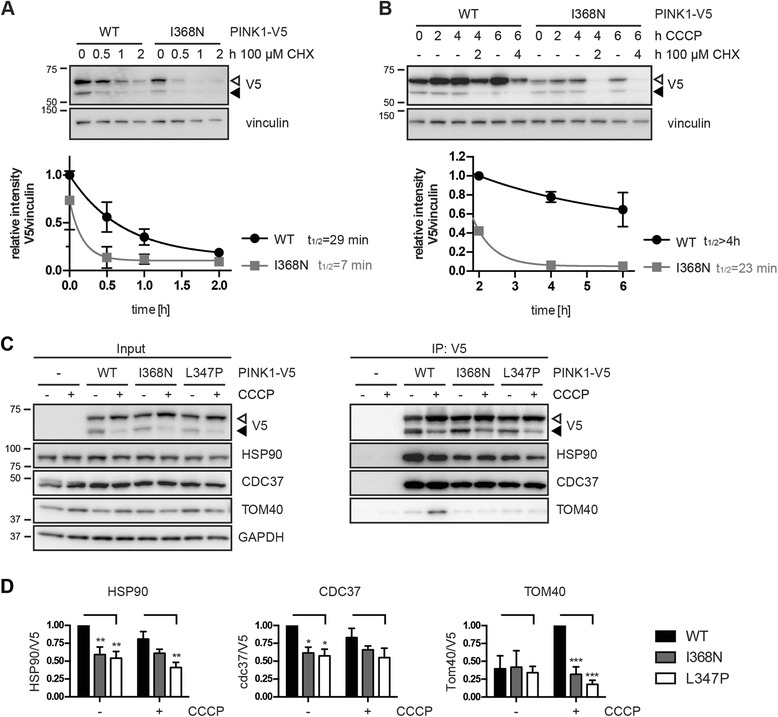
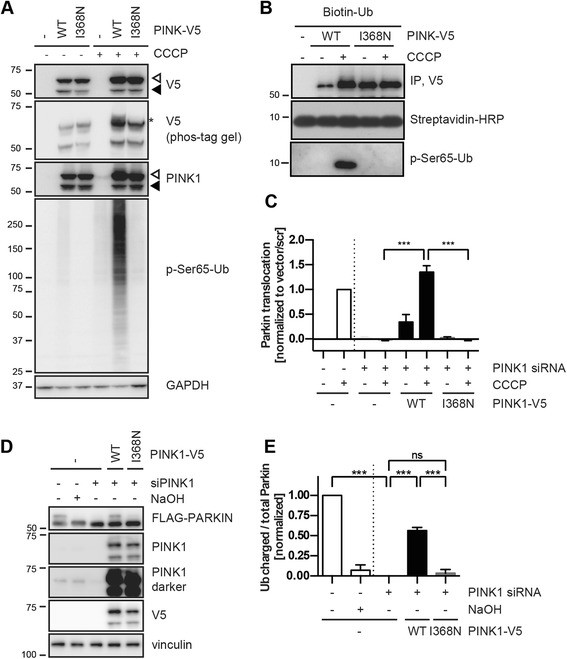
Results: Under endogenous conditions, PINK1 p.I368N is expressed, imported, and N-terminally processed in healthy mitochondria similar to PINK1 wild type (WT). Upon mitochondrial damage, however, full-length PINK1 p.I368N is not sufficiently stabilized on the outer mitochondrial membrane (OMM) resulting in loss of mitochondrial quality control. We found that binding of PINK1 p.I368N to the co-chaperone complex HSP90/CDC37 is reduced and stress-induced interaction with TOM40 of the mitochondrial protein import machinery is abolished. Analysis of a structural PINK1 p.I368N model additionally suggested impairments of Ub kinase activity as the ATP-binding pocket was found deformed and the substrate Ub was slightly misaligned within the active site of the kinase. Functional assays confirmed the lack of Ub kinase activity.
Conclusions: Here we demonstrated that mutant PINK1 p.I368N can not be stabilized on the OMM upon mitochondrial stress and due to conformational changes in the active site does not exert kinase activity towards Ub. In patients' fibroblasts, biochemical assays and by structural analyses, we unraveled two pathomechanisms that lead to loss of function upon mutation of p.I368N and highlight potential strategies for future drug development.
Keywords: Autophagy; E3 ubiquitin ligase; Mitochondria; Mitophagy; PARK2; PARKIN; PINK1; Parkinson’s disease; Phospho-ubiquitin; Ubiquitin.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
