

The evolution of lifespan and age-dependent cancer risk
- PMID: 28439564
- PMCID: PMC5400291
- DOI: 10.1016/j.trecan.2016.09.004
The evolution of lifespan and age-dependent cancer risk
Abstract
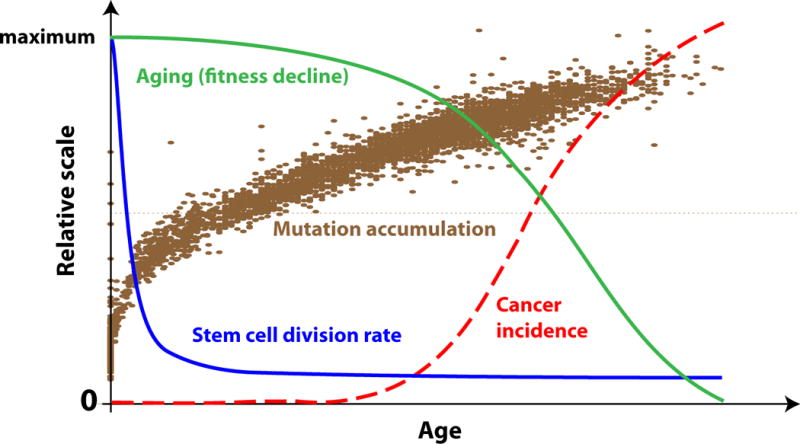
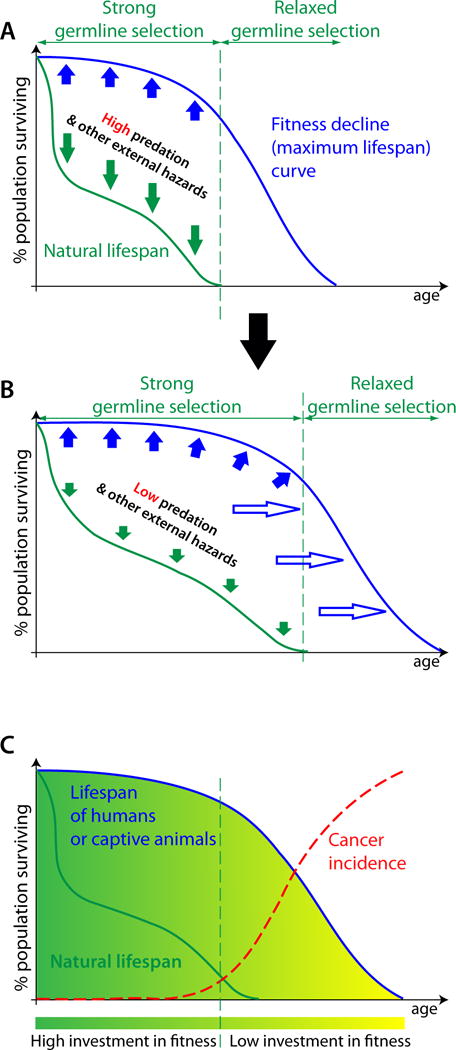
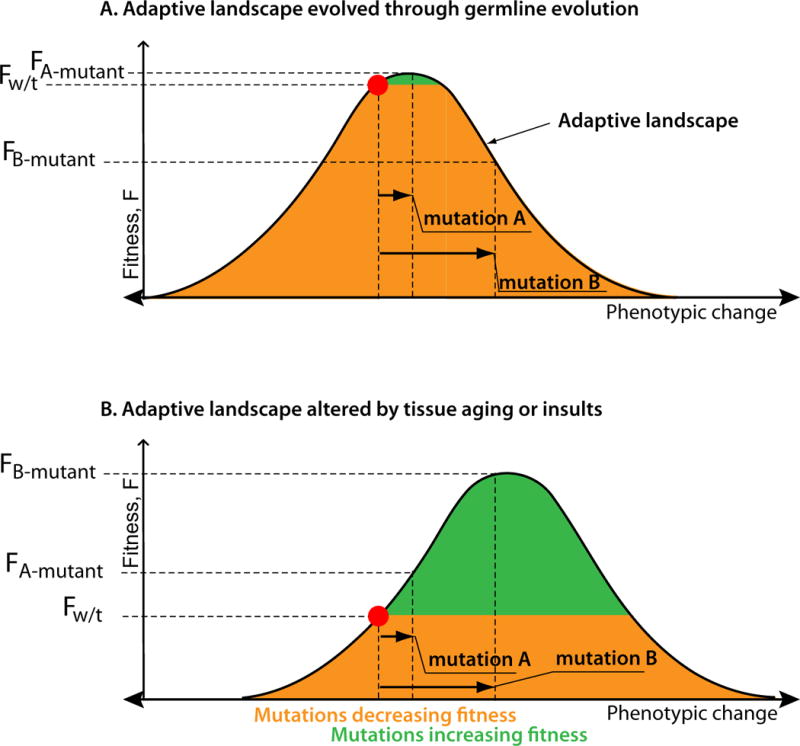
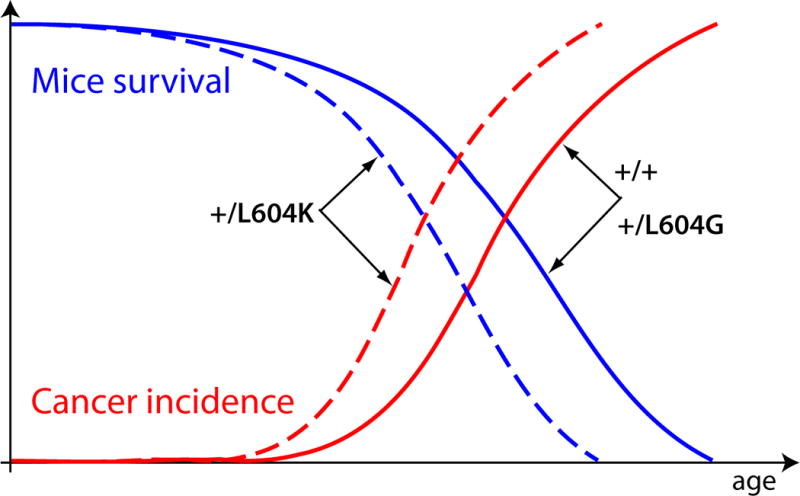
The Armitage-Doll multi-stage model of carcinogenesis tremendously refocused cancer science by postulating that carcinogenesis is driven by a sequence of genetic changes in cells. Age-dependent cancer incidence thus has been explained in terms of the time necessary for oncogenic mutations to occur. While the multi-step nature of cancer evolution is well-supported by evidence, the mutation-centric theory is unable to explain a number of phenomena, such as the disproportion between cancer frequency and animal body size or the scaling of cancer incidence to animal lifespan. In this paper, we present a theoretical review of the current paradigm and discuss some fundamental evolutionary theory postulates that explain why cancer incidence is a function of lifespan and physiological, not chronological, aging.
Figures
References
-
- Nowell P, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497. - PubMed
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. - PubMed
-
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical