

Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation
- PMID: 28441135
- PMCID: PMC5404927
- DOI: 10.7554/eLife.24463
Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation
Erratum in
-
Correction: Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation.Elife. 2019 Aug 5;8:e50707. doi: 10.7554/eLife.50707. Elife. 2019. PMID: 31378216 Free PMC article.
Abstract
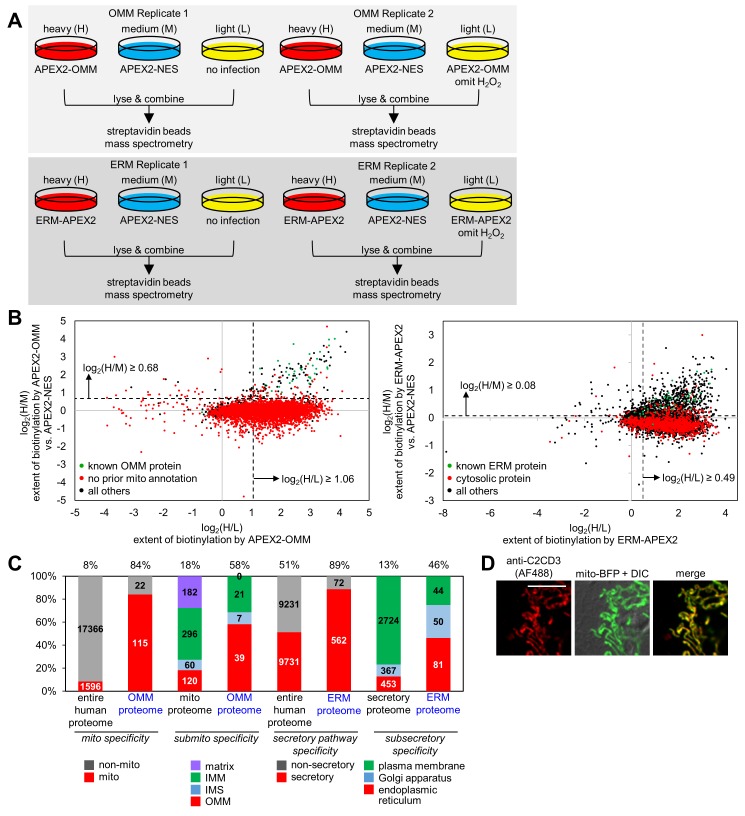
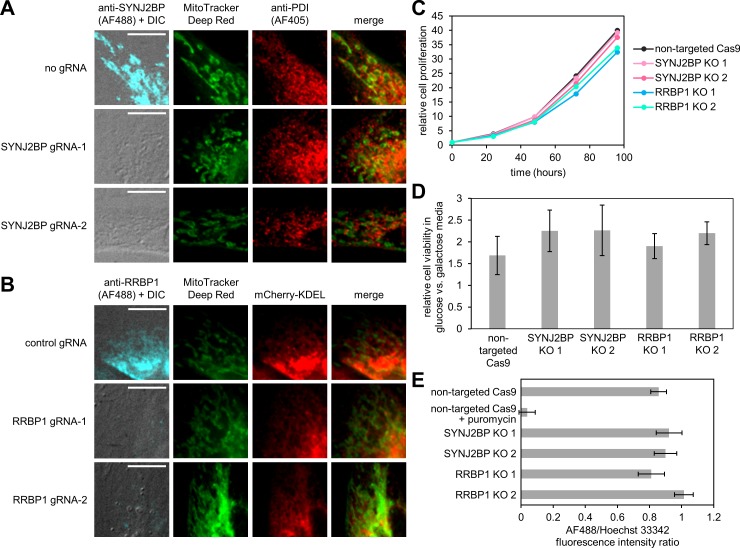
The cytosol-facing membranes of cellular organelles contain proteins that enable signal transduction, regulation of morphology and trafficking, protein import and export, and other specialized processes. Discovery of these proteins by traditional biochemical fractionation can be plagued with contaminants and loss of key components. Using peroxidase-mediated proximity biotinylation, we captured and identified endogenous proteins on the outer mitochondrial membrane (OMM) and endoplasmic reticulum membrane (ERM) of living human fibroblasts. The proteomes of 137 and 634 proteins, respectively, are highly specific and highlight 94 potentially novel mitochondrial or ER proteins. Dataset intersection identified protein candidates potentially localized to mitochondria-ER contact sites. We found that one candidate, the tail-anchored, PDZ-domain-containing OMM protein SYNJ2BP, dramatically increases mitochondrial contacts with rough ER when overexpressed. Immunoprecipitation-mass spectrometry identified ribosome-binding protein 1 (RRBP1) as SYNJ2BP's ERM binding partner. Our results highlight the power of proximity biotinylation to yield insights into the molecular composition and function of intracellular membranes.
Keywords: APEX2; biochemistry; cell biology; human; microscopy; mitochondria-ER junctions; mitochondria-associated membrane; promiscuous enzymatic labeling; subcellular regions.
Conflict of interest statement
No competing interests declared.
Listed as an inventor on a patent on peroxidase-mediated proteomic mapping technology (US Application No. 61/497155).
Figures








References
-
- Adam MG, Berger C, Feldner A, Yang WJ, Wüstehube-Lausch J, Herberich SE, Pinder M, Gesierich S, Hammes HP, Augustin HG, Fischer A. Synaptojanin-2 binding protein stabilizes the notch ligands DLL1 and DLL4 and inhibits sprouting angiogenesis. Circulation Research. 2013;113:1206–1218. doi: 10.1161/CIRCRESAHA.113.301686. - DOI - PubMed
-
- Ahn K, Szczesna-Skorupa E, Kemper B. The amino-terminal 29 amino acids of cytochrome P450 2c1 are sufficient for retention in the endoplasmic reticulum. The Journal of Biological Chemistry. 1993;268:18726–18733. - PubMed
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. the gene ontology consortium. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. - DOI - PMC - PubMed
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. 1995;57::289–300. doi: 10.2307/2346101. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
