

Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans
- PMID: 28444186
- PMCID: PMC6251520
- DOI: 10.1093/hmg/ddx141
Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans
Abstract
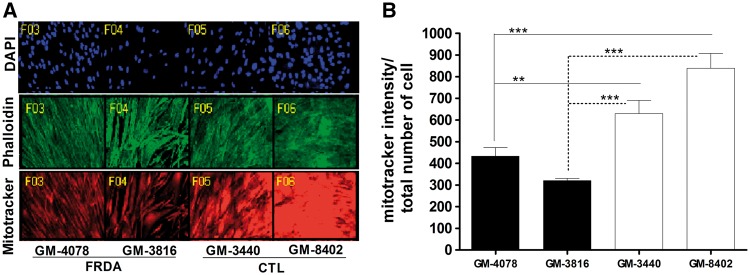
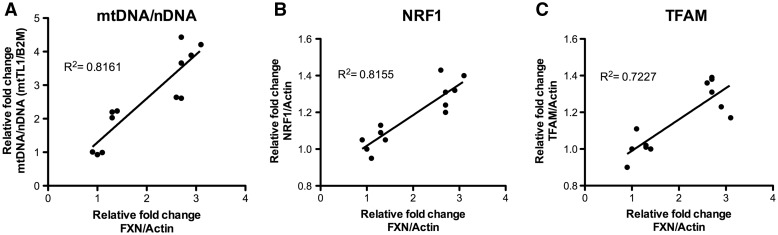
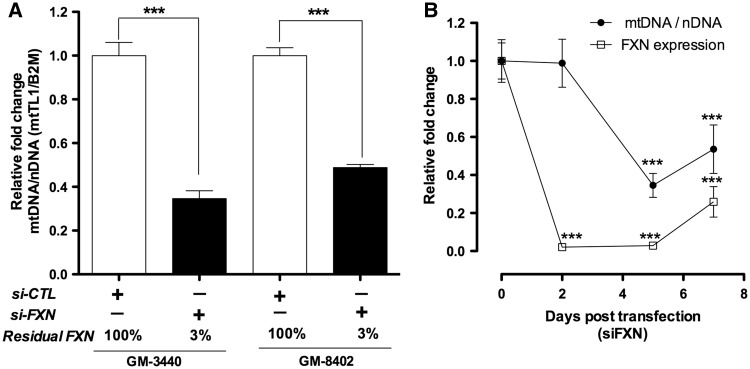
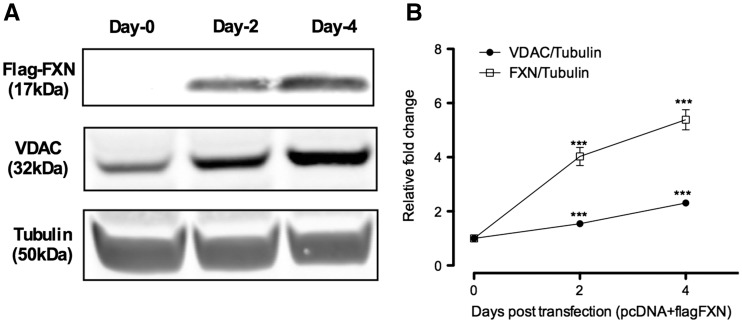
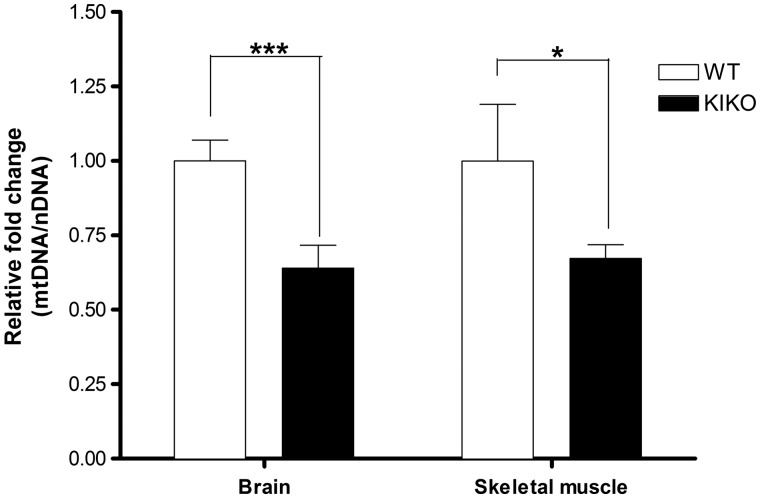
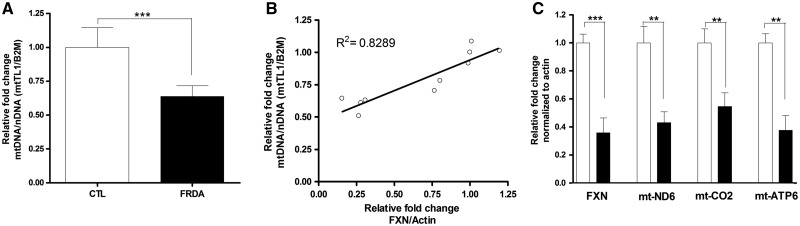
Friedreich's ataxia (FRDA) is a neurodegenerative disease caused by inherited deficiency of the mitochondrial protein Frataxin (FXN), which has no approved therapy and is an area in which biomarkers are needed for clinical development. Here, we investigated the consequences of FXN deficiency in patient-derived FRDA fibroblast cell models, the FRDA mouse model KIKO, and in whole blood collected from patients with FRDA. We observed decreased mitochondrial copy number in all the three FRDA models tested: cells, mice and patient blood. In addition, we observed 40% residual mitochondrial gene expression in FRDA patient blood. These deficiencies of mitochondrial biogenesis in FRDA cells and patient blood are significantly correlated with FXN expression, consistent with the idea that the decreased mitochondrial biogenesis is a consequence of FXN deficiency. The observations appear relevant to the FRDA pathophysiological mechanism, as FXN-dependent deficiency in mitochondrial biogenesis and consequent mitochondrial bioenergetic defect could contribute to the neurodegenerative process. The observations may also have translational potential, as mitochondrial biogenesis could now be followed as a clinical biomarker of FRDA as a correlate of disease severity, progression, and therapeutic effect. Also, mitochondrial copy number in blood is objective, scalar and more investigator-independent than clinical-neurological patient rating scales. Thus, FXN deficiency causes mitochondrial deficiency in FRDA cells, the KIKO mouse model, and in whole blood of patients with FRDA, and this deficiency could potentially be used in clinical trial design.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Koutnikova H., Campuzano V., Foury F., Dollé P., Cazzalini O., Koenig M. (1997) Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet., 16, 345–351. - PubMed
-
- Bradley J., Blake J., Chamberlain S., Thomas P., Cooper J., Schapira A. (2000) Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet., 9, 275–282. - PubMed
-
- Campuzano V., Montermini L., Moltò M.D., Pianese L. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science, 271, 1423. - PubMed
-
- Parkinson M.H., Boesch S., Nachbauer W., Mariotti C., Giunti P. (2013) Clinical features of Friedreich's ataxia: classical and atypical phenotypes. J. Neurochem., 126, 103–117. - PubMed
-
- Vankan P. (2013) Prevalence gradients of Friedreich's Ataxia and R1b haplotype in Europe co‐localize, suggesting a common Palaeolithic origin in the Franco‐Cantabrian ice age refuge. J. Neurochem., 126, 11–20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
