

Systematic Epigenomic Analysis Reveals Chromatin States Associated with Melanoma Progression
- PMID: 28445736
- PMCID: PMC5473172
- DOI: 10.1016/j.celrep.2017.03.078
Systematic Epigenomic Analysis Reveals Chromatin States Associated with Melanoma Progression
Abstract
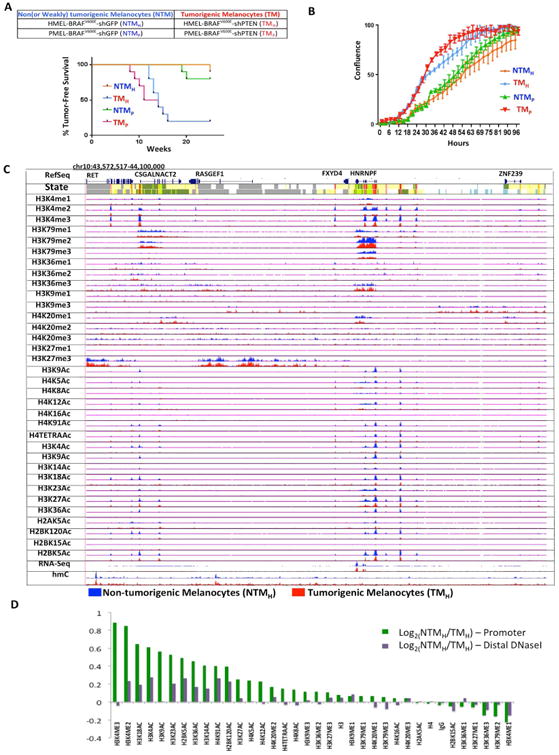
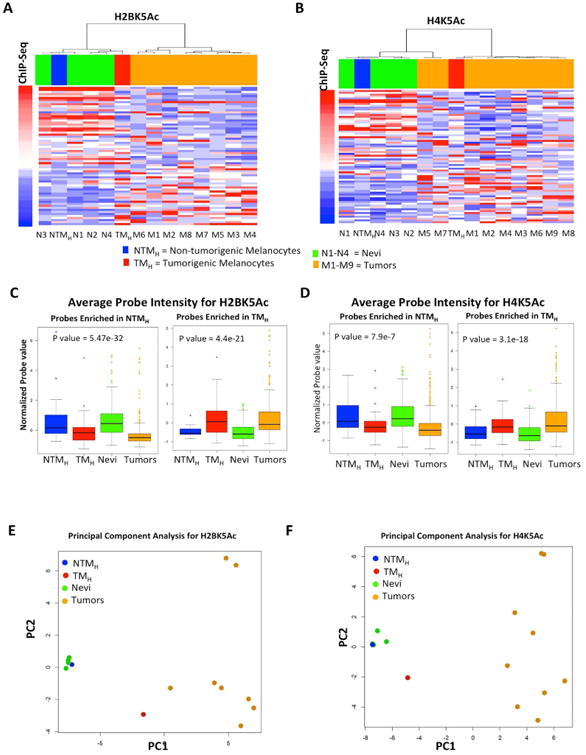
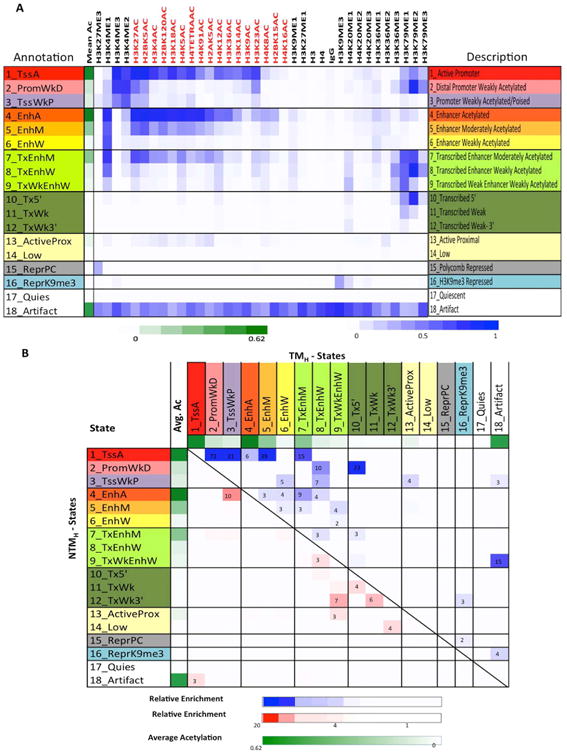
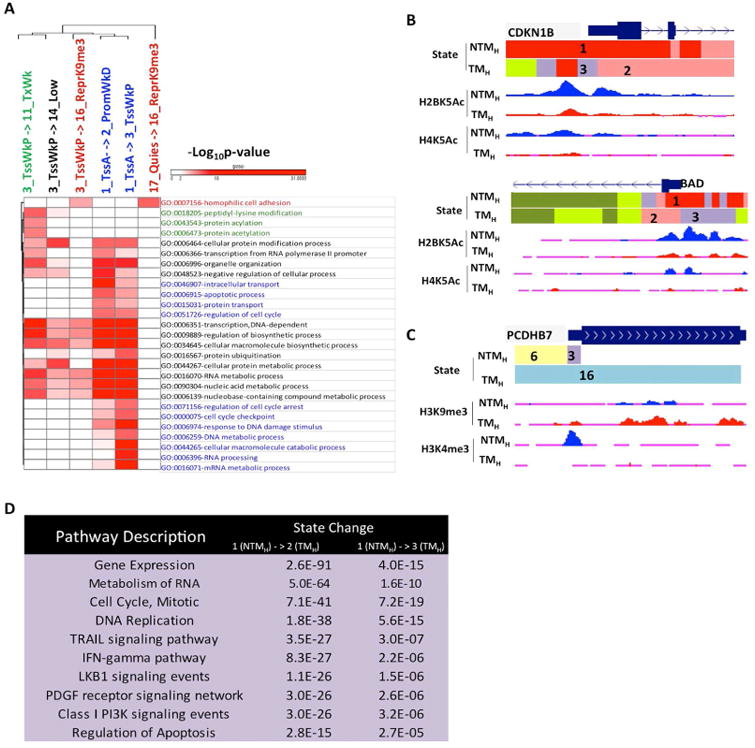
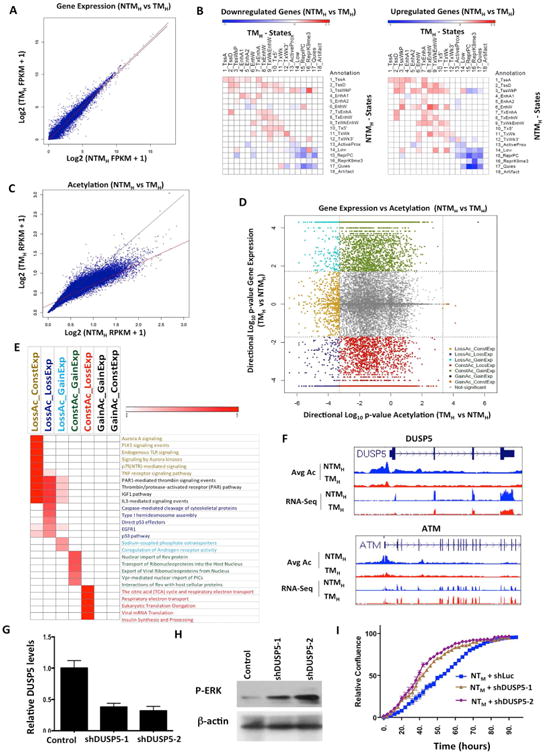
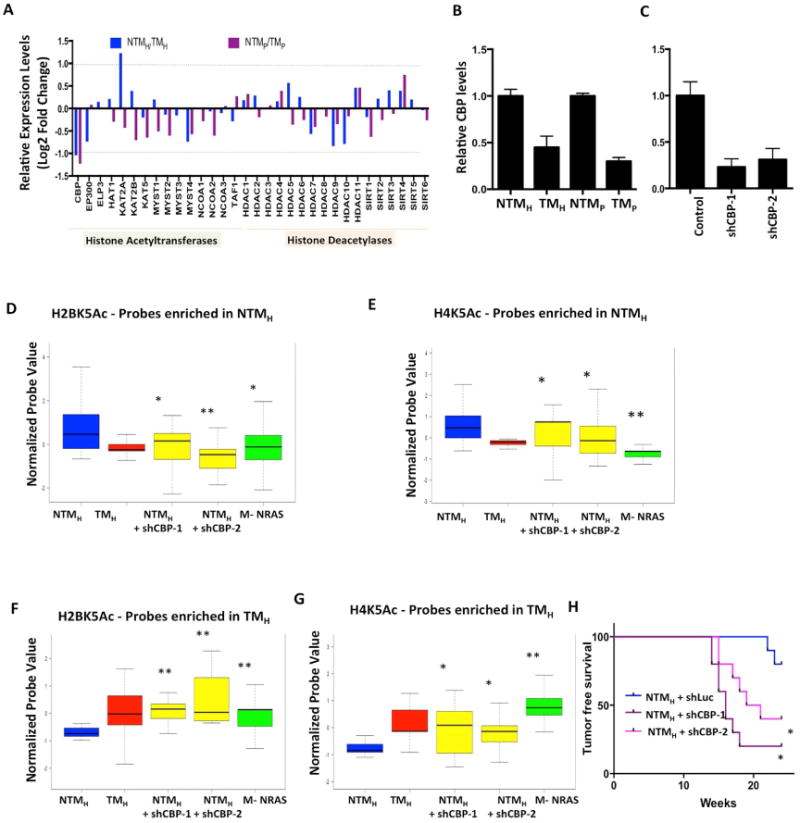
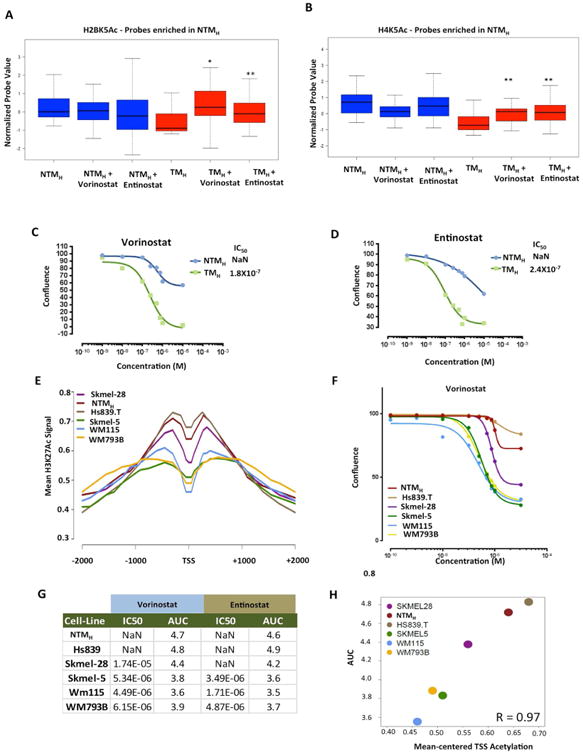
The extent and nature of epigenomic changes associated with melanoma progression is poorly understood. Through systematic epigenomic profiling of 35 epigenetic modifications and transcriptomic analysis, we define chromatin state changes associated with melanomagenesis by using a cell phenotypic model of non-tumorigenic and tumorigenic states. Computation of specific chromatin state transitions showed loss of histone acetylations and H3K4me2/3 on regulatory regions proximal to specific cancer-regulatory genes in important melanoma-driving cell signaling pathways. Importantly, such acetylation changes were also observed between benign nevi and malignant melanoma human tissues. Intriguingly, only a small fraction of chromatin state transitions correlated with expected changes in gene expression patterns. Restoration of acetylation levels on deacetylated loci by histone deacetylase (HDAC) inhibitors selectively blocked excessive proliferation in tumorigenic cells and human melanoma cells, suggesting functional roles of observed chromatin state transitions in driving hyperproliferative phenotype. Through these results, we define functionally relevant chromatin states associated with melanoma progression.
Keywords: CBP; ChIP-seq; DUSP5; HDAC; chromatin state; epigenome; histone modifications; melanoma.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Bennett DC. Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res. 2016;29:122–140. - PubMed
-
- Busse A, Keilholz U. Role of TGF-beta in melanoma. Curr Pharm Biotechnol. 2011;12:2165–2175. - PubMed
-
- Campioni M, Santini D, Tonini G, Murace R, Dragonetti E, Spugnini EP, Baldi A. Role of Apaf-1, a key regulator of apoptosis, in melanoma progression and chemoresistance. Exp Dermatol. 2005;14:811–818. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
