

PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability
- PMID: 28446439
- PMCID: PMC7802885
- DOI: 10.1158/2159-8290.CD-16-1297
PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability
Abstract
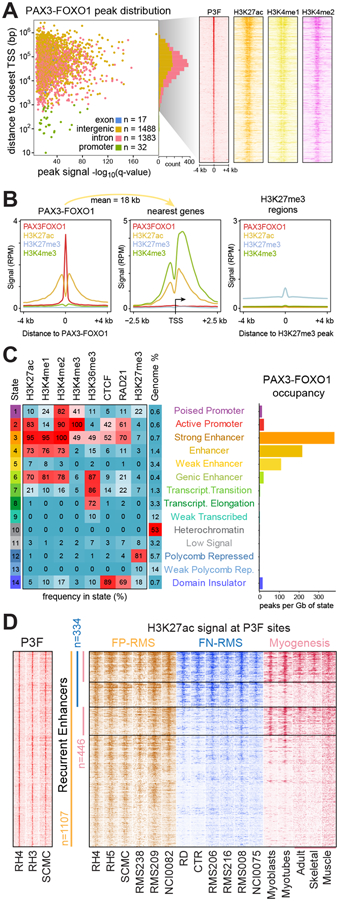
Alveolar rhabdomyosarcoma is a life-threatening myogenic cancer of children and adolescent young adults, driven primarily by the chimeric transcription factor PAX3-FOXO1. The mechanisms by which PAX3-FOXO1 dysregulates chromatin are unknown. We find PAX3-FOXO1 reprograms the cis-regulatory landscape by inducing de novo super enhancers. PAX3-FOXO1 uses super enhancers to set up autoregulatory loops in collaboration with the master transcription factors MYOG, MYOD, and MYCN. This myogenic super enhancer circuitry is consistent across cell lines and primary tumors. Cells harboring the fusion gene are selectively sensitive to small-molecule inhibition of protein targets induced by, or bound to, PAX3-FOXO1-occupied super enhancers. Furthermore, PAX3-FOXO1 recruits and requires the BET bromodomain protein BRD4 to function at super enhancers, resulting in a complete dependence on BRD4 and a significant susceptibility to BRD inhibition. These results yield insights into the epigenetic functions of PAX3-FOXO1 and reveal a specific vulnerability that can be exploited for precision therapy.Significance: PAX3-FOXO1 drives pediatric fusion-positive rhabdomyosarcoma, and its chromatin-level functions are critical to understanding its oncogenic activity. We find that PAX3-FOXO1 establishes a myoblastic super enhancer landscape and creates a profound subtype-unique dependence on BET bromodomains, the inhibition of which ablates PAX3-FOXO1 function, providing a mechanistic rationale for exploring BET inhibitors for patients bearing PAX-fusion rhabdomyosarcoma. Cancer Discov; 7(8); 884-99. ©2017 AACR.This article is highlighted in the In This Issue feature, p. 783.
©2017 American Association for Cancer Research.
Conflict of interest statement
All authors declare no conflicts of interest.
Figures






References
-
- Rabbitts T Chromosomal translocations in human cancer. Nature. 1994;372(6502):143–9. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
