

Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration
- PMID: 28446513
- PMCID: PMC5786429
- DOI: 10.1136/jmedgenet-2017-104540
Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration
Abstract
Background: Variation in the ABCA4 gene is causal for, or associated with, a wide range of phenotypes from early onset Mendelian retinal dystrophies to late-onset complex disorders such as age-related macular degeneration (AMD). Despite substantial progress in determining the causal genetic variation, even complete sequencing of the entire open reading frame and splice sites of ABCA4 identifies biallelic mutations in only 60%-70% of cases; 20%-25% remain with one mutation and no mutations are found in 10%-15% of cases with clinically confirmed ABCA4 disease. This study was designed to identify missing causal variants specifically in monoallelic cases of ABCA4 disease.
Methods: Direct sequencing and analysis were performed in a large familial ABCA4 disease cohort of predominately European descent (n=643). Patient phenotypes were assessed from clinical and retinal imaging data.
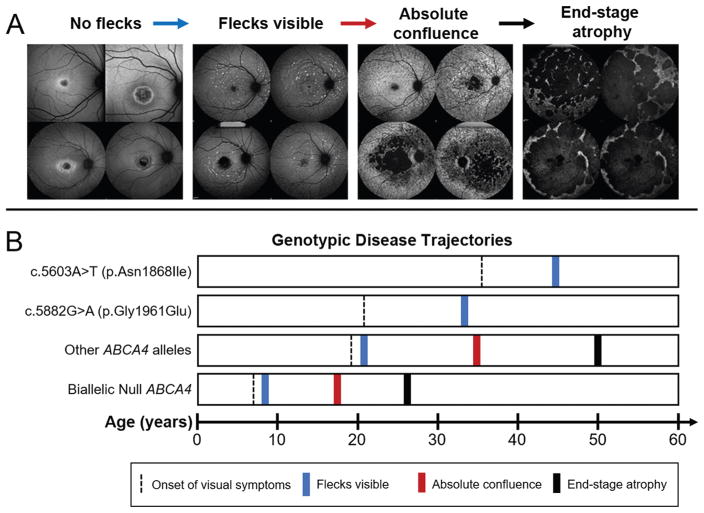
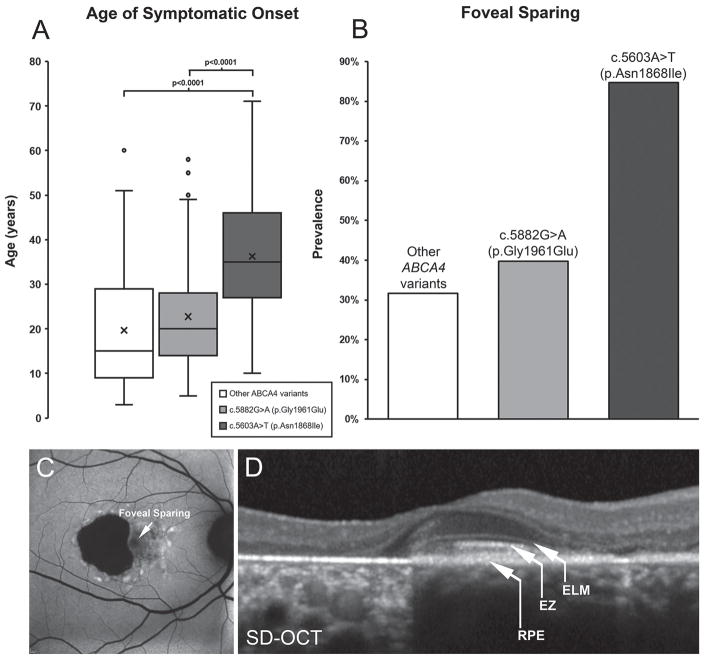
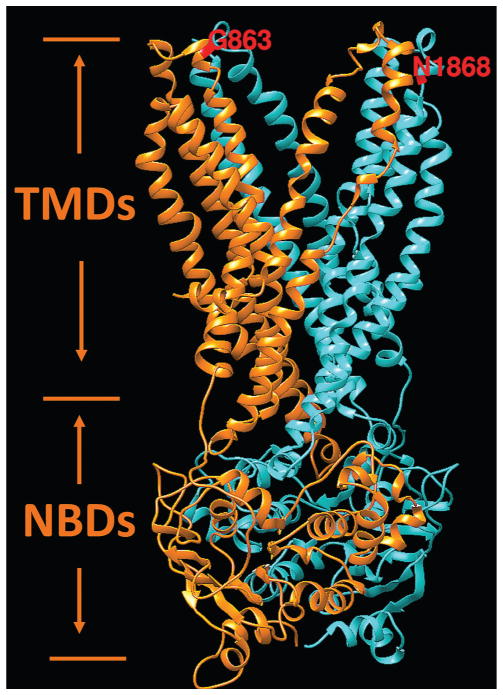
Results: We determined that a hypomorphic ABCA4 variant c.5603A>T (p.Asn1868Ile), previously considered benign due to high minor allele frequency (MAF) (~7%) in the general population, accounts for 10% of the disease, >50% of the missing causal alleles in monoallelic cases, ~80% of late-onset cases and distinguishes ABCA4 disease from AMD. It results in a distinct clinical phenotype characterised by late-onset of symptoms (4th decade) and foveal sparing (85%). Intragenic modifying effects involving this variant and another, c.2588G>C (p.Gly863Ala) allele, were also identified.
Conclusions: These findings substantiate the causality of frequent missense variants and their phenotypic outcomes as a significant contribution to ABCA4 disease, particularly the late-onset phenotype, and its clinical variation. They also suggest a significant revision of diagnostic screening and assessment of ABCA4 variation in aetiology of retinal diseases.
Keywords: ABCA4; Age-related macular degeneration; Stargardt disease; foveal sparing; hypomorphic variant.
© Article author(s) (or their employer(s) unless otherwise stated in the text of the article) 2017. All rights reserved. No commercial use is permitted unless otherwise expressly granted.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. - PubMed
-
- Cremers FP, van de Pol DJ, van Driel M, den Hollander AI, van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Deutman AF, Hoyng CB. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–62. - PubMed
-
- Martínez-Mir A, Paloma E, Allikmets R, Ayuso C, del Rio T, Dean M, Vilageliu L, Gonzàlez-Duarte R, Balcells S. Retinitis Pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18:11–12. - PubMed
-
- Shroyer NF, Lewis RA, Yatsenko AN, Lupski JR. Null missense ABCR (ABCA4) mutations in a family with stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2001;42:2757–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical