

The histone variant H2A.Z promotes efficient cotranscriptional splicing in S. cerevisiae
- PMID: 28446598
- PMCID: PMC5411710
- DOI: 10.1101/gad.295188.116
The histone variant H2A.Z promotes efficient cotranscriptional splicing in S. cerevisiae
Abstract
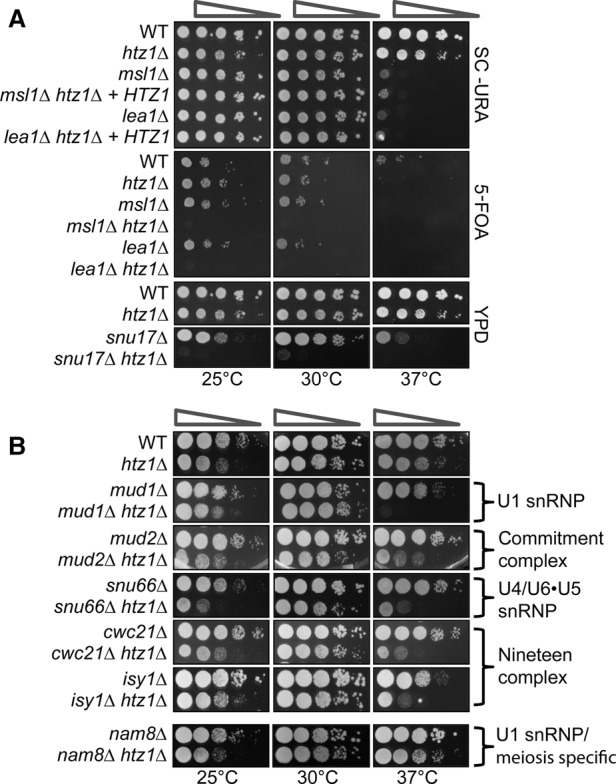
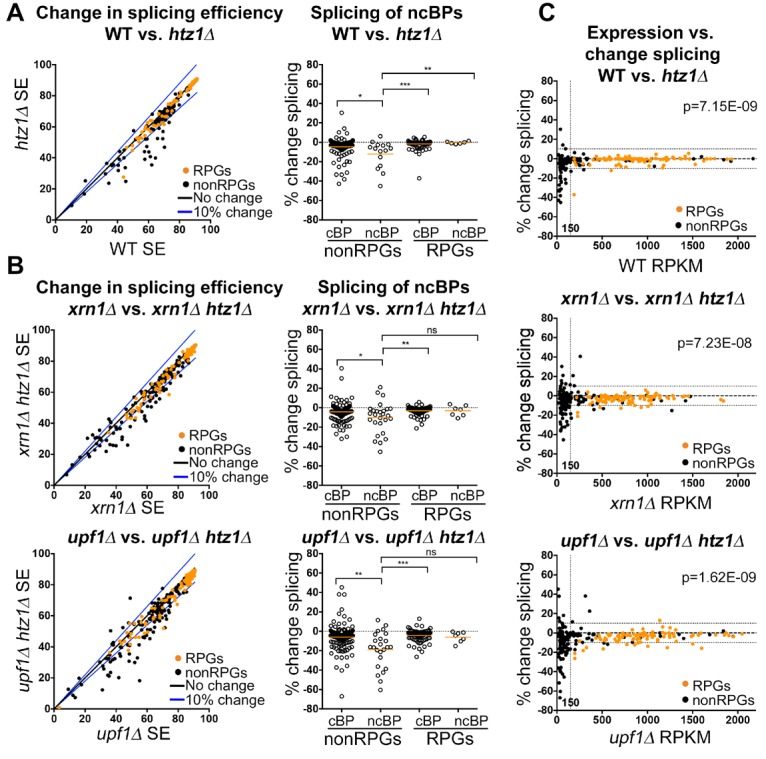
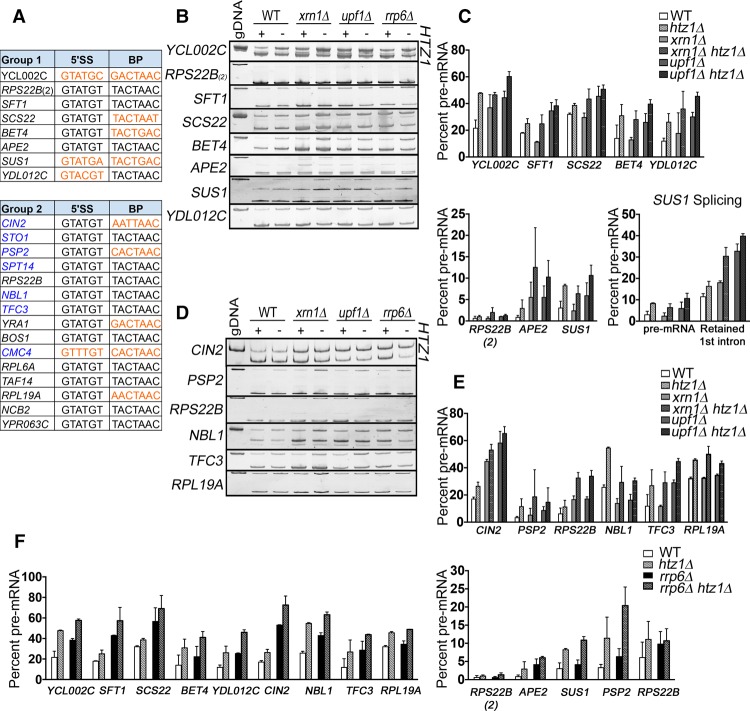
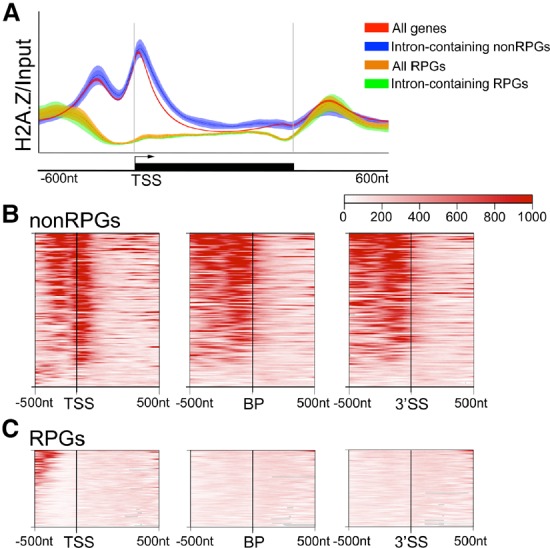
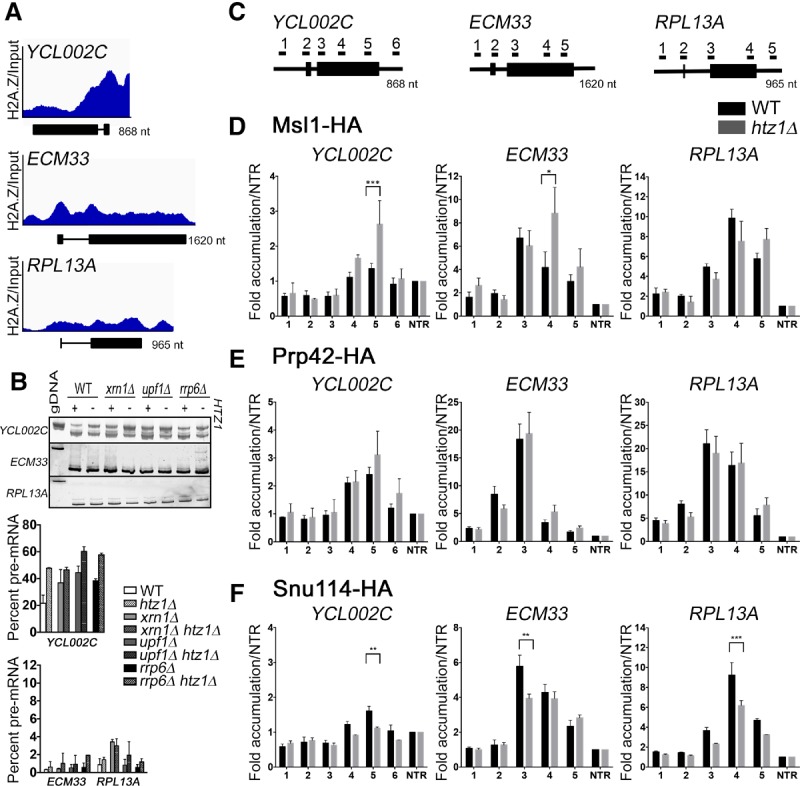
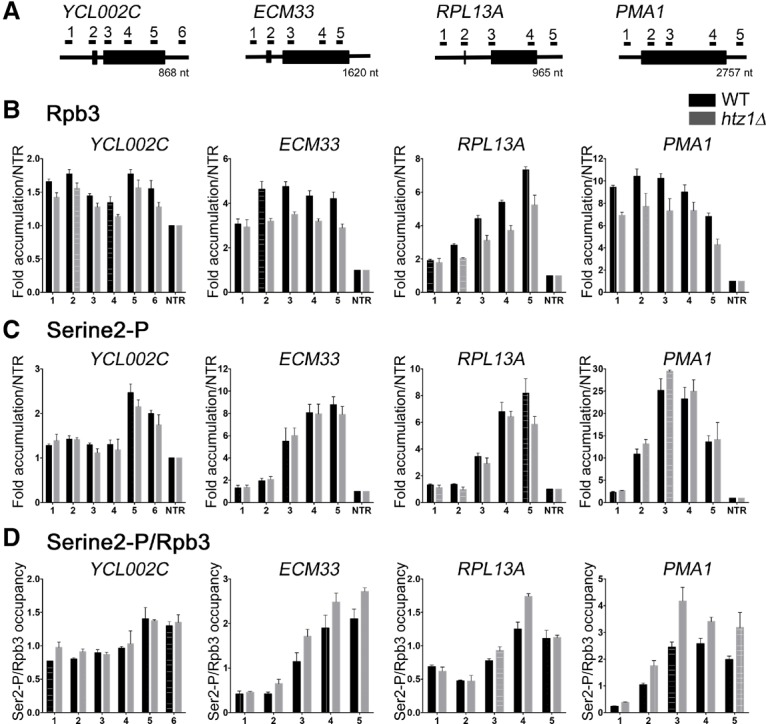
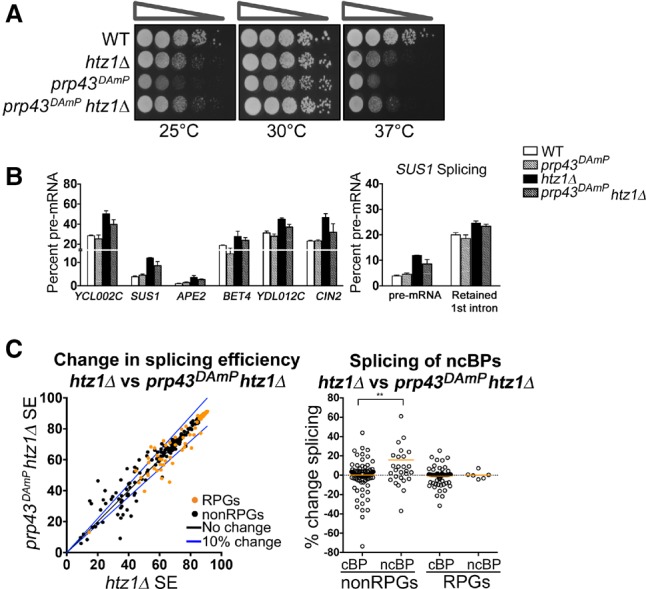
In eukaryotes, a dynamic ribonucleic protein machine known as the spliceosome catalyzes the removal of introns from premessenger RNA (pre-mRNA). Recent studies show the processes of RNA synthesis and RNA processing to be spatio-temporally coordinated, indicating that RNA splicing takes place in the context of chromatin. H2A.Z is a highly conserved histone variant of the canonical histone H2A. In Saccharomyces cerevisiae, H2A.Z is deposited into chromatin by the SWR-C complex, is found near the 5' ends of protein-coding genes, and has been implicated in transcription regulation. Here we show that splicing of intron-containing genes in cells lacking H2A.Z is impaired, particularly under suboptimal splicing conditions. Cells lacking H2A.Z are especially dependent on a functional U2 snRNP (small nuclear RNA [snRNA] plus associated proteins), as H2A.Z shows extensive genetic interactions with U2 snRNP-associated proteins, and RNA sequencing (RNA-seq) reveals that introns with nonconsensus branch points are particularly sensitive to H2A.Z loss. Consistently, H2A.Z promotes efficient spliceosomal rearrangements involving the U2 snRNP, as H2A.Z loss results in persistent U2 snRNP association and decreased recruitment of downstream snRNPs to nascent RNA. H2A.Z impairs transcription elongation, suggesting that spliceosome rearrangements are tied to H2A.Z's role in elongation. Depletion of disassembly factor Prp43 suppresses H2A.Z-mediated splice defects, indicating that, in the absence of H2A.Z, stalled spliceosomes are disassembled, and unspliced RNAs are released. Together, these data demonstrate that H2A.Z is required for efficient pre-mRNA splicing and indicate a role for H2A.Z in coordinating the kinetics of transcription elongation and splicing.
Keywords: H2A.Z; HTZ1; RNA processing; Swr1; budding yeast; chromatin; pre-mRNA splicing.
© 2017 Neves et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
The histone variant H2A.Z promotes splicing of weak introns.Genes Dev. 2017 Apr 1;31(7):688-701. doi: 10.1101/gad.295287.116. Genes Dev. 2017. PMID: 28446597 Free PMC article.
-
The splicing factor Prp17 interacts with the U2, U5 and U6 snRNPs and associates with the spliceosome pre- and post-catalysis.Biochem J. 2008 Dec 15;416(3):365-74. doi: 10.1042/BJ20081195. Biochem J. 2008. PMID: 18691155
-
Efficient RNA polymerase II pause release requires U2 snRNP function.Mol Cell. 2021 May 6;81(9):1920-1934.e9. doi: 10.1016/j.molcel.2021.02.016. Epub 2021 Mar 8. Mol Cell. 2021. PMID: 33689748
-
The specificity of H2A.Z occupancy in the yeast genome and its relationship to transcription.Curr Genet. 2020 Oct;66(5):939-944. doi: 10.1007/s00294-020-01087-7. Epub 2020 Jun 14. Curr Genet. 2020. PMID: 32537667 Review.
-
Branch site recognition by the spliceosome.RNA. 2024 Oct 16;30(11):1397-1407. doi: 10.1261/rna.080198.124. RNA. 2024. PMID: 39187383 Free PMC article. Review.
Cited by
-
The TORC1 activates Rpd3L complex to deacetylate Ino80 and H2A.Z and repress autophagy.Sci Adv. 2023 Mar 10;9(10):eade8312. doi: 10.1126/sciadv.ade8312. Epub 2023 Mar 8. Sci Adv. 2023. PMID: 36888706 Free PMC article.
-
Histone Marks-Dependent Effect on Alternative Splicing: New Perspectives for Targeted Splicing Modulation in Cancer?Int J Mol Sci. 2022 Jul 27;23(15):8304. doi: 10.3390/ijms23158304. Int J Mol Sci. 2022. PMID: 35955433 Free PMC article. Review.
-
Evolution of the Early Spliceosomal Complex-From Constitutive to Regulated Splicing.Int J Mol Sci. 2021 Nov 18;22(22):12444. doi: 10.3390/ijms222212444. Int J Mol Sci. 2021. PMID: 34830325 Free PMC article. Review.
-
Competitive Chemical Reaction Kinetic Model of Nucleosome Assembly Using the Histone Variant H2A.Z and H2A In Vitro.Int J Mol Sci. 2023 Oct 31;24(21):15846. doi: 10.3390/ijms242115846. Int J Mol Sci. 2023. PMID: 37958827 Free PMC article.
-
Mathematical modeling identifies potential gene structure determinants of co-transcriptional control of alternative pre-mRNA splicing.Nucleic Acids Res. 2018 Nov 16;46(20):10598-10607. doi: 10.1093/nar/gky870. Nucleic Acids Res. 2018. PMID: 30272246 Free PMC article.
References
-
- Amberg DC, Burke D, Strathern J. 2005. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
-
- Amit M, Donyo M, Hollander D, Goren A, Kim E, Gelfman S, Lev-Maor G, Burstein D, Schwartz S, Postolsky B, et al. 2012. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep 1: 543–556. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases