

Cystic fibrosis: current therapeutic targets and future approaches
- PMID: 28449677
- PMCID: PMC5408469
- DOI: 10.1186/s12967-017-1193-9
Cystic fibrosis: current therapeutic targets and future approaches
Abstract
Objectives: Study of currently approved drugs and exploration of future clinical development pipeline therapeutics for cystic fibrosis, and possible limitations in their use.
Methods: Extensive literature search using individual and a combination of key words related to cystic fibrosis therapeutics.
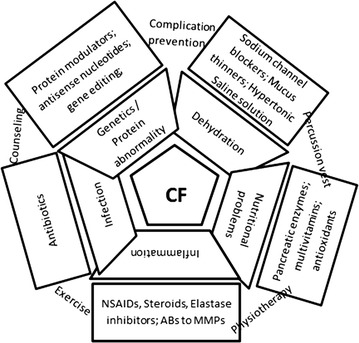
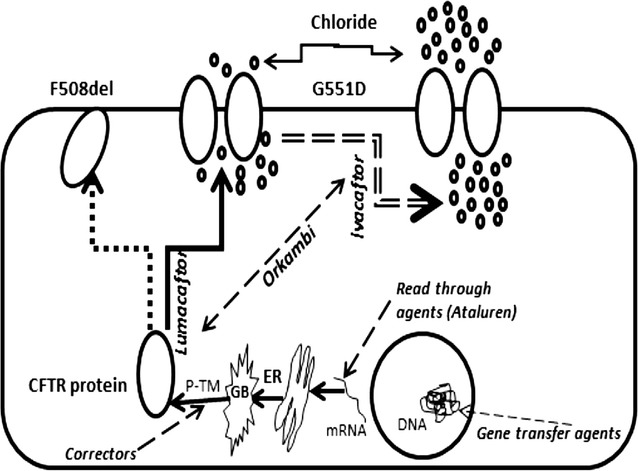
Key findings: Cystic fibrosis is an autosomal recessive disorder due to mutations in CFTR gene leading to abnormality of chloride channels in mucus and sweat producing cells. Respiratory system and GIT are primarily involved but eventually multiple organs are affected leading to life threatening complications. Management requires drug therapy, extensive physiotherapy and nutritional support. Previously, the focus was on symptomatic improvement and complication prevention but recently the protein rectifiers are being studied which are claimed to correct underlying structural and functional abnormalities. Some improvement is observed by the corrector drugs. Other promising approaches are gene therapy, targeting of cellular interactomes, and newer drugs for symptomatic improvement.
Conclusions: The treatment has a long way to go as most of the existing therapeutics is for older children. Other limiting factors include mutation class, genetic profile, drug interactions, adverse effects, and cost. Novel approaches like gene transfer/gene editing, disease modeling and search for alternative targets are warranted.
Keywords: CFTR; Chloride; Hereditary; Respiratory; Sweat.
Figures
References
-
- Johnson LG, Boyles SE, Wilson J, Boucher RC. Normalization of raised sodium absorption and raised calcium-mediated chloride secretion by adenovirus-mediated expression of cystic fibrosis transmembrane conductance regulator in primary human cystic fibrosis airway epithelial cells. J Clin Invest. 1995;95:1377–1382. doi: 10.1172/JCI117789. - DOI - PMC - PubMed
-
- Cystic fibrosis foundation patient registry: annual data report to the center directors, 2014. https://www.cff.org/2014_CFF_Annual_Data_Report_to_the_Center_Directors..... Accessed 11 Mar 2016.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
