

Congenital Adrenal Hyperplasia
- PMID: 28450075
- PMCID: PMC5624825
- DOI: 10.1016/j.jpag.2017.04.001
Congenital Adrenal Hyperplasia
Abstract
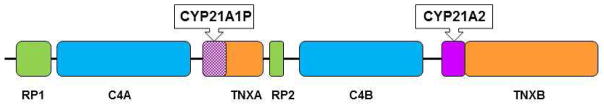
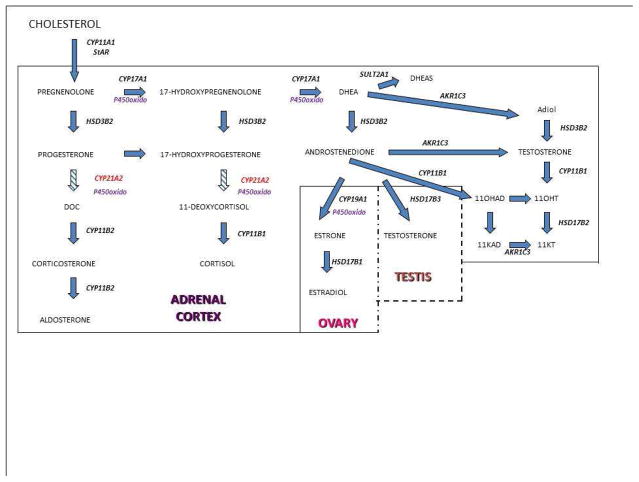
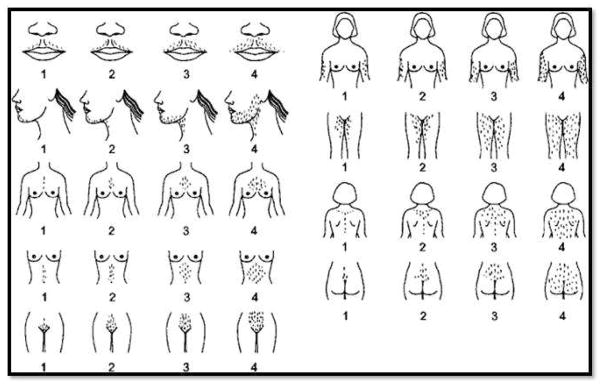
The congenital adrenal hyperplasias comprise a family of autosomal recessive disorders that disrupt adrenal steroidogenesis. The most common form is due to 21-hydroxylase deficiency associated with mutations in the 21-hydroxylase gene, which is located at chromosome 6p21. The clinical features associated with each disorder of adrenal steroidogenesis represent a clinical spectrum that reflect the consequences of the specific mutations. Treatment goals include normal linear growth velocity and "on-time" puberty in affected children. For adolescent and adult women, treatment goals include regularization of menses, prevention of progression of hirsutism, and preservation of fertility. For adolescent and adult men, prevention and early treatment of testicular adrenal rest tumors is beneficial. In this article key aspects regarding pathophysiology, diagnosis, and treatment of congenital adrenal hyperplasia are reviewed.
Keywords: Ambiguous genitalia; CYP21A2; Congenital adrenal hyperplasia; Hyperandrogenism; Premature adrenarche; Premature pubarche.
Copyright © 2017 North American Society for Pediatric and Adolescent Gynecology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Delle Piane L, Rinaudo PF, Miller WL. 150 years of congenital adrenal hyperplasia: translation and commentary of De Crecchio’s classic paper from 1865. Endocrinology. 2015;156:1210. - PubMed
-
- Decourt MJ, Jayle MF, Baulieu E. Virilisme cliniquement tardif avec excretion de pregnanetriol et insuffisance de la production du cortisol. Ann Endocrinol (Paris) 1957;18:416. - PubMed
-
- Thil’en A, Nordenstrom A, Hagenfeldt L, von Dobeln U, Guthenberg C, Larsson A. Benefits of neonatal screening for congenital adrenal hyperplasia (21-hydroxylase deficiency) in Sweden. Pediatrics. 1998;101(4):E11. - PubMed
-
- Therrell BL, Jr, Berenbaum SA, Manter-Kapanke V, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101(4 Pt 1):583. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
