

An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition
- PMID: 28450382
- PMCID: PMC5460994
- DOI: 10.1084/jem.20160855
An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition
Abstract
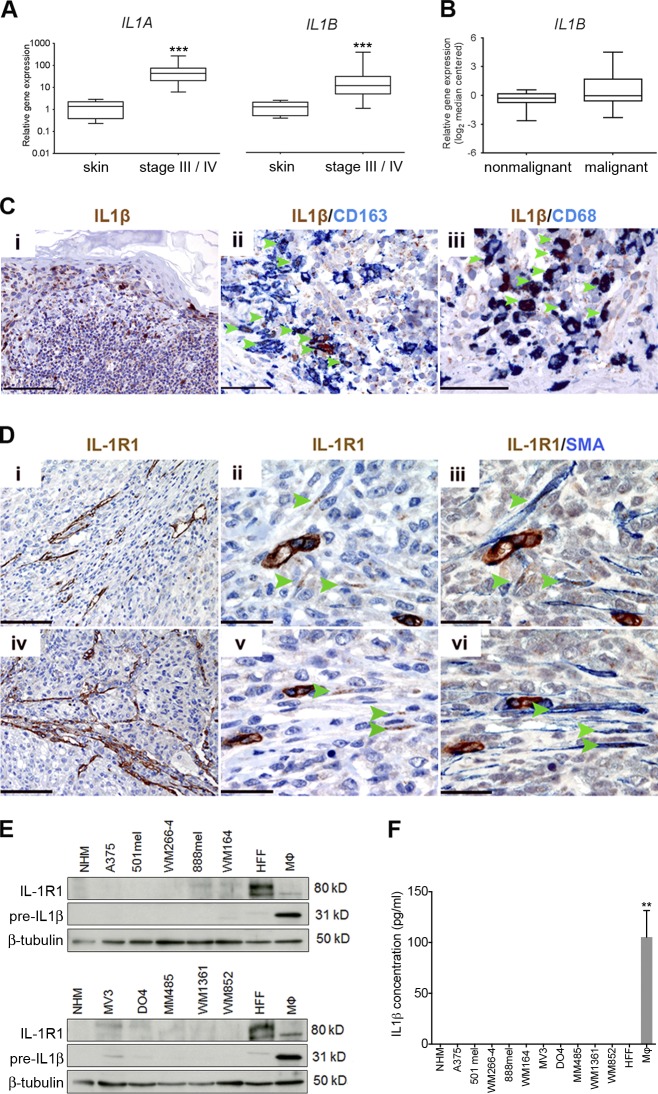
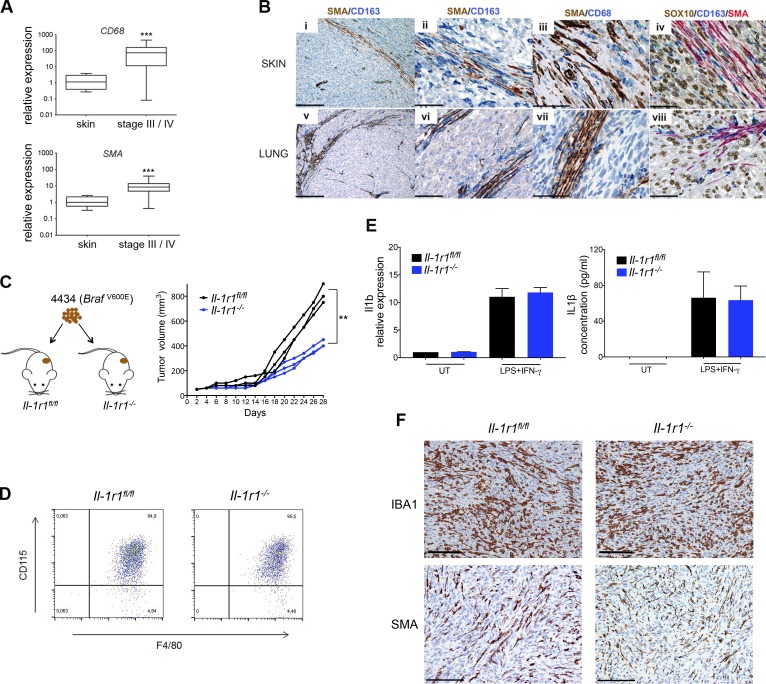
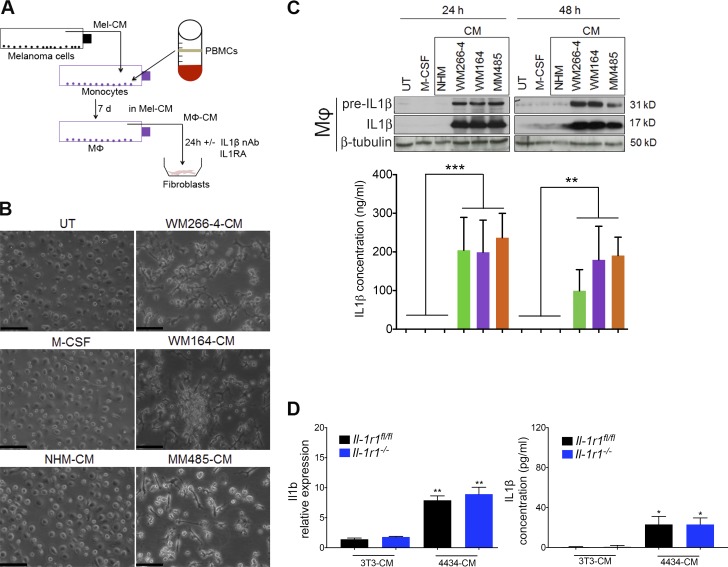
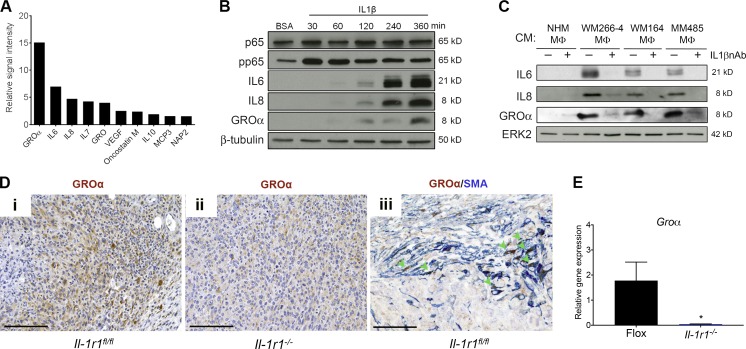
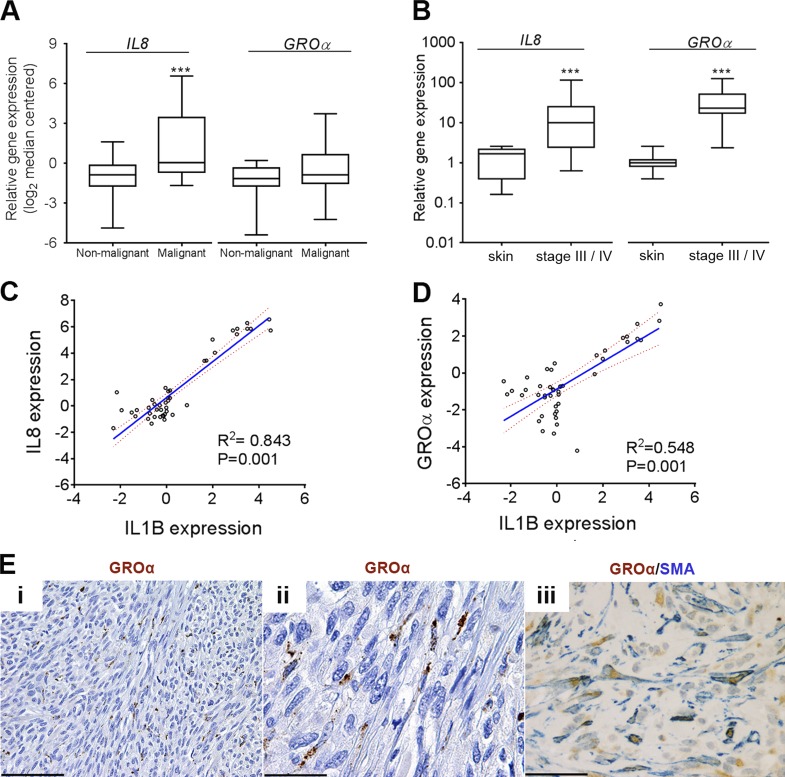
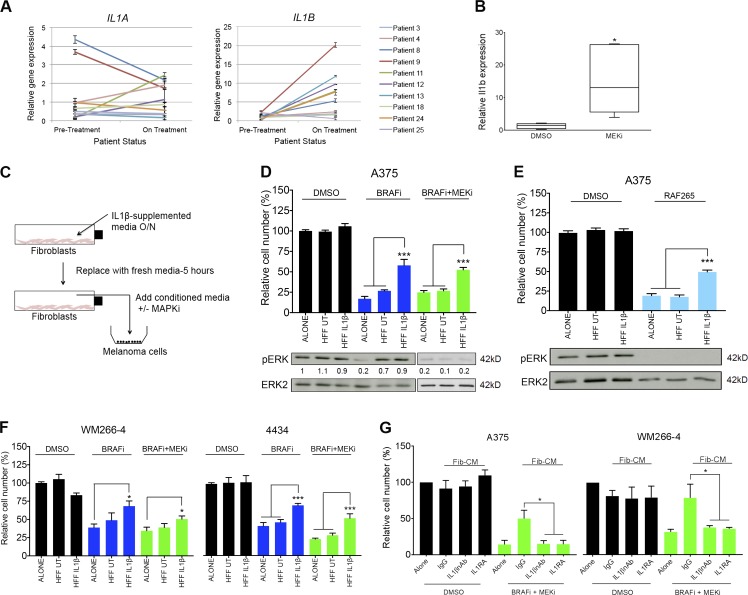
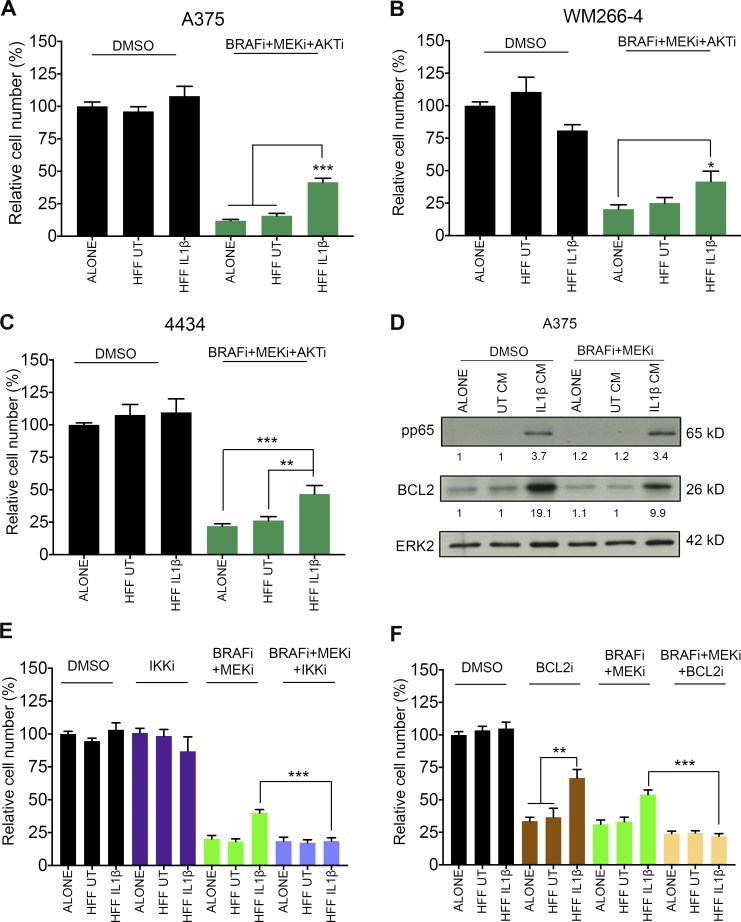
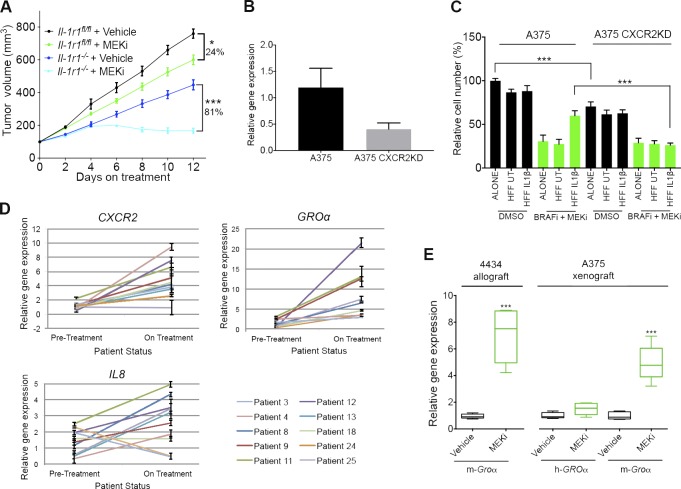
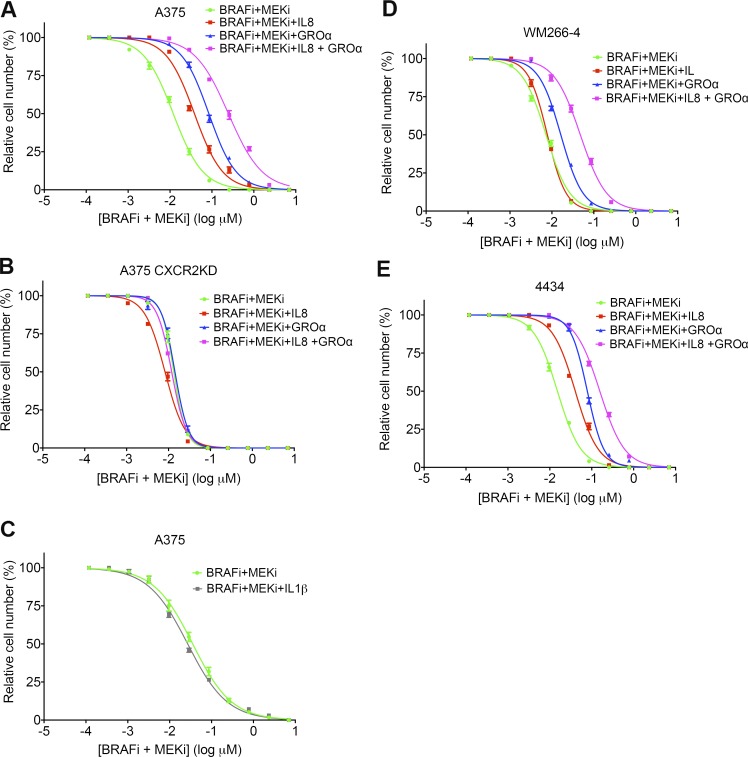
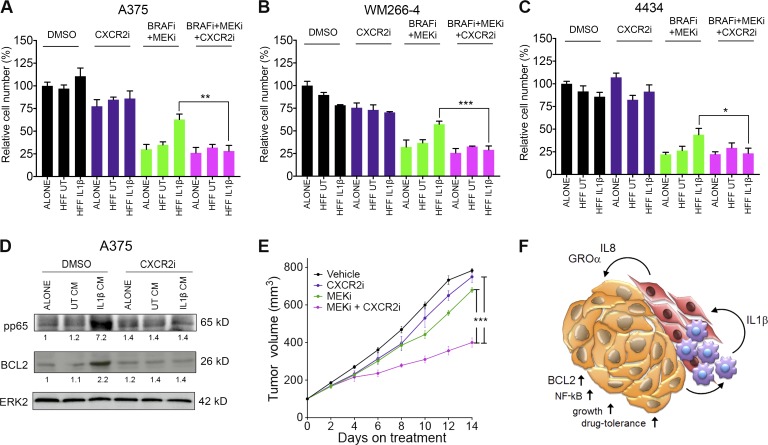
Mitogen-activated protein kinase (MAPK) pathway antagonists induce profound clinical responses in advanced cutaneous melanoma, but complete remissions are frustrated by the development of acquired resistance. Before resistance emerges, adaptive responses establish a mutation-independent drug tolerance. Antagonizing these adaptive responses could improve drug effects, thereby thwarting the emergence of acquired resistance. In this study, we reveal that inflammatory niches consisting of tumor-associated macrophages and fibroblasts contribute to treatment tolerance through a cytokine-signaling network that involves macrophage-derived IL-1β and fibroblast-derived CXCR2 ligands. Fibroblasts require IL-1β to produce CXCR2 ligands, and loss of host IL-1R signaling in vivo reduces melanoma growth. In tumors from patients on treatment, signaling from inflammatory niches is amplified in the presence of MAPK inhibitors. Signaling from inflammatory niches counteracts combined BRAF/MEK (MAPK/extracellular signal-regulated kinase kinase) inhibitor treatment, and consequently, inhibiting IL-1R or CXCR2 signaling in vivo enhanced the efficacy of MAPK inhibitors. We conclude that melanoma inflammatory niches adapt to and confer drug tolerance toward BRAF and MEK inhibitors early during treatment.
© 2017 Young et al.
Figures
References
-
- Abdulaal W.H., Walker C.R., Costello R., Redondo-Castro E., Mufazalov I.A., Papaemmanouil A., Rothwell N.J., Allan S.M., Waisman A., Pinteaux E., and Müller W.. 2016. Characterization of a conditional interleukin-1 receptor 1 mouse mutant using the Cre/LoxP system. Eur. J. Immunol. 46:912–918. 10.1002/eji.201546075 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
