

The Design of the Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) Trial
- PMID: 28454798
- PMCID: PMC5586211
- DOI: 10.1016/j.ahj.2017.02.008
The Design of the Valsartan for Attenuating Disease Evolution in Early Sarcomeric Hypertrophic Cardiomyopathy (VANISH) Trial
Abstract
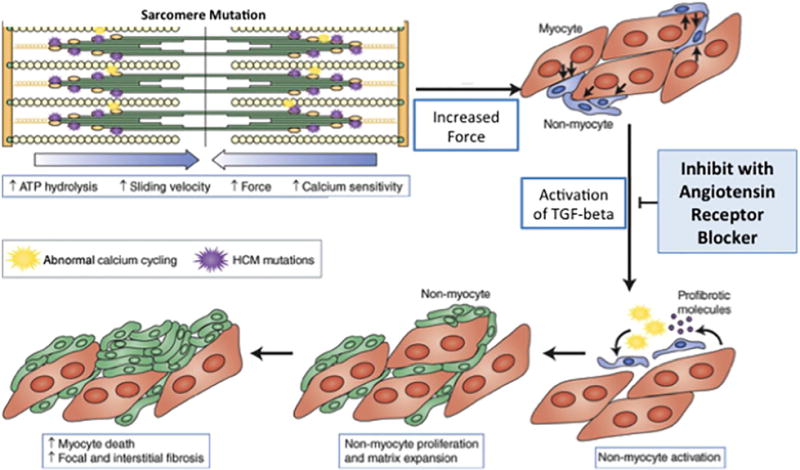
Hypertrophic cardiomyopathy (HCM) is often caused by sarcomere gene mutations, resulting in left ventricular hypertrophy (LVH), myocardial fibrosis, and increased risk of sudden cardiac death and heart failure. Studies in mouse models of sarcomeric HCM demonstrated that early treatment with an angiotensin receptor blocker (ARB) reduced development of LVH and fibrosis. In contrast, prior human studies using ARBs for HCM have targeted heterogeneous adult cohorts with well-established disease. The VANISH trial is testing the safety and feasibility of disease-modifying therapy with an ARB in genotyped HCM patients with early disease.
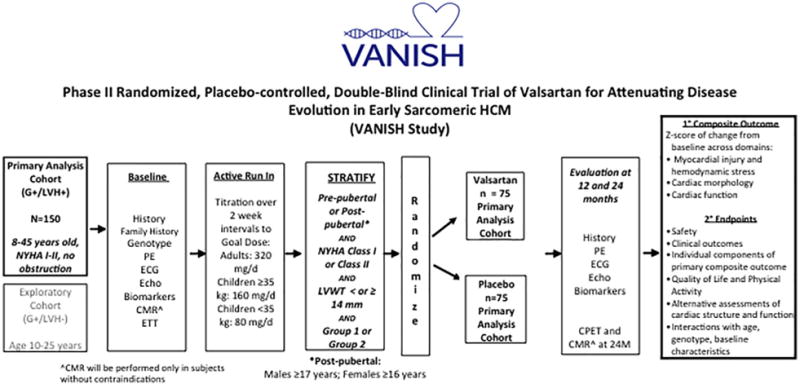
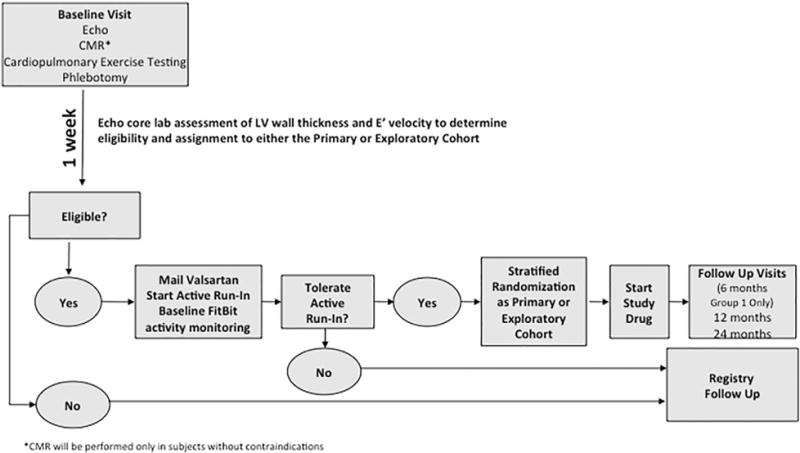
Methods: A randomized, placebo-controlled, double-blind clinical trial is being conducted in sarcomere mutation carriers, 8 to 45 years old, with HCM and no/minimal symptoms, or those with early phenotypic manifestations but no LVH. Participants are randomly assigned to receive valsartan 80 to 320 mg daily (depending on age and weight) or placebo. The primary endpoint is a composite of 9 z-scores in domains representing myocardial injury/hemodynamic stress, cardiac morphology, and function. Total z-scores reflecting change from baseline to final visits will be compared between treatment groups. Secondary endpoints will assess the impact of treatment on mutation carriers without LVH, and analyze the influence of age, sex, and genotype.
Conclusions: The VANISH trial is testing a new strategy of disease modification for treating sarcomere mutation carriers with early HCM, and those at risk for its development. In addition, further insight into disease mechanisms, response to therapy, and phenotypic evolution will be gained.
Copyright © 2017. Published by Elsevier Inc.
Conflict of interest statement
Conflict of Interest: NONE.
Figures
References
-
- Maron BJ, Gardin JM, Flack JM, et al. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults-Echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation. 1995;92:785–9. - PubMed
-
- Maron BJ, Casey SA, Hauser RG, et al. Clinical course of hypertrophic cardiomyopathy with survival to advanced age. J Am Coll Cardiol. 2003;42:882–8. - PubMed
-
- Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687–713. - PubMed
-
- Gersh BJ, Maron BJ, Bonow RO, et al. ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;2011 - PubMed
-
- Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064–75. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
