

Heterogeneous Phenotypes in Lipid Storage Myopathy Due to ETFDH Gene Mutations
- PMID: 28456887
- PMCID: PMC5874208
- DOI: 10.1007/8904_2017_27
Heterogeneous Phenotypes in Lipid Storage Myopathy Due to ETFDH Gene Mutations
Abstract
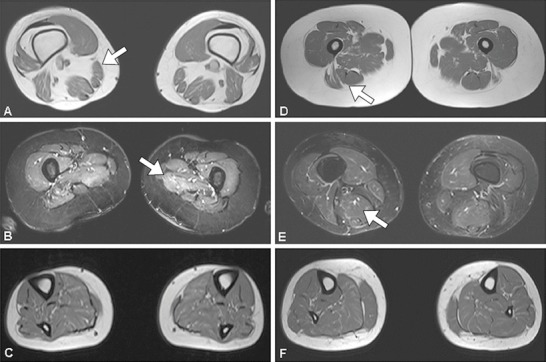
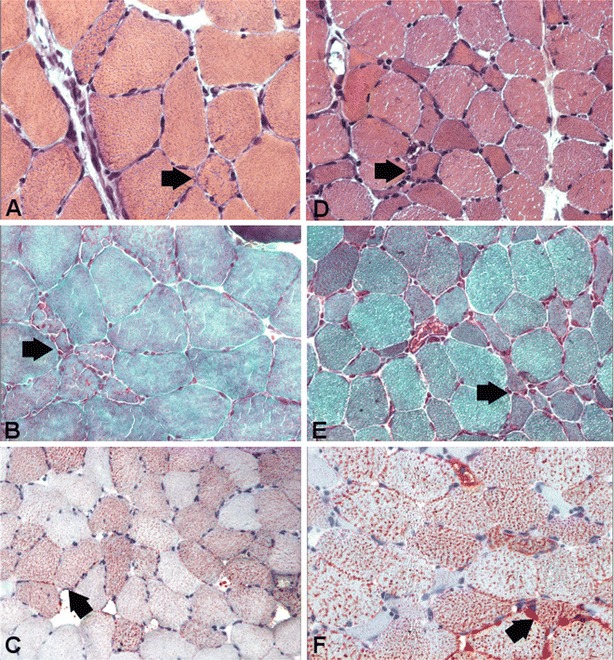
We present six novel patients affected by lipid storage myopathy (LSM) presenting mutations in the ETFDH gene. Although the diagnosis of multiple acyl-coenzyme-A dehydrogenase deficiency (MADD) in adult life is difficult, it is rewarding because of the possibility of treating patients with carnitine or riboflavin, leading to a full recovery. In our patients, a combination of precipitating risk factors including previous anorexia, alcoholism, poor nutrition, and pregnancy contributed to a metabolic critical condition that precipitated the catabolic state.In the present series of cases, five novel mutations have been identified in the ETFDH gene. We propose clinical guidelines to screen patients with LSM due to different defects, in order to obtain a fast diagnosis and offer appropriate treatment. In such patients, early diagnosis and treatment as well as avoiding risk factors are part of clinical management.Specific biochemical studies are indicated to identify the type of LSM, such as level of free carnitine and acyl-carnitines and studies or organic acidemia. Indeed, when a patient is biochemically diagnosed with secondary carnitine deficiency, a follow-up with appropriate clinical-molecular protocol and genetic analysis is important to establish the final diagnosis, since riboflavin can be supplemented with benefit if riboflavin-responsive MADD is present. In muscle biopsies, increased lipophagy associated with p62-positive aggregates was observed. The clinical improvement can be attributed to the removal of an autophagic block, which appears to be reversible in this LSM.
Figures
References
-
- Angelini C (2017) Metabolites-mitochondria-macrophages (MMM): new therapeutic avenues for inflammation and muscle atrophy. Transl Cancer Res 5(4):S8–S11 doi: 10.21037/tcr.2017.01.37
-
- Freneaux E, Sheffield VC, Molin L, Shires A, Rhead WJ. Glutaric acidemia type II: heterogeneity in beta-oxidation flux, polypeptide synthesis, and complementary DNA mutations in the alpha-subunit of electron transfer flavoprotein in eight patients. J Clin Invest. 1992;90:1679–1686. doi: 10.1172/JCI116040. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
