

Transmission patterns and evolution of respiratory syncytial virus in a community outbreak identified by genomic analysis
- PMID: 28458916
- PMCID: PMC5399923
- DOI: 10.1093/ve/vex006
Transmission patterns and evolution of respiratory syncytial virus in a community outbreak identified by genomic analysis
Abstract
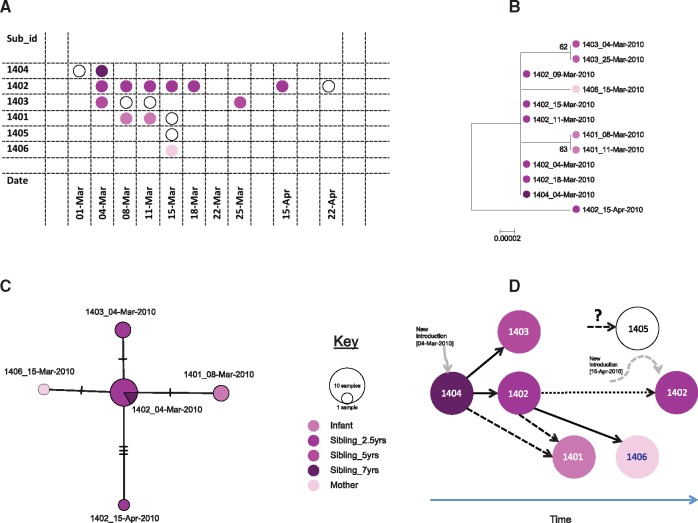
Detailed information on the source, spread and evolution of respiratory syncytial virus (RSV) during seasonal community outbreaks remains sparse. Molecular analyses of attachment (G) gene sequences from hospitalized cases suggest that multiple genotypes and variants co-circulate during epidemics and that RSV persistence over successive seasons is characterized by replacement and multiple new introductions of variants. No studies have defined the patterns of introduction, spread and evolution of RSV at the local community and household level. We present a whole genome sequence analysis of 131 RSV group A viruses collected during 6-month household-based RSV infection surveillance in Coastal Kenya, 2010 within an area of 12 km2. RSV infections were identified by regular symptom-independent screening of all household members twice weekly. Phylogenetic analysis revealed that the RSV A viruses in nine households were closely related to genotype GA2 and fell within a single branch of the global phylogeny. Genomic analysis allowed the detection of household-specific variation in seven households. For comparison, using only G gene analysis, household-specific variation was found only in one of the nine households. Nucleotide changes were observed both intra-host (viruses identified from same individual in follow-up sampling) and inter-host (viruses identified from different household members) and these coupled with sampling dates enabled a partial reconstruction of the within household transmission chains. The genomic evolutionary rate for the household dataset was estimated as 2.307 × 10 - 3 (95% highest posterior density: 0.935-4.165× 10 - 3) substitutions/site/year. We conclude that (i) at the household level, most RSV infections arise from the introduction of a single virus variant followed by accumulation of household specific variation and (ii) analysis of complete virus genomes is crucial to better understand viral transmission in the community. A key question arising is whether prevention of RSV introduction or spread within the household by vaccinating key transmitting household members would lead to a reduced onward community-wide transmission.
Keywords: RSV; WAIFW; community transmission; full-genome sequencing; household transmission.
Figures




References
-
- Cane P. A. (2001) ‘Molecular Epidemiology of Respiratory Syncytial Virus’, Reviews in Medical Virology, 11/2: 103–16. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
