

Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia
- PMID: 28459997
- PMCID: PMC6402316
- DOI: 10.1093/brain/awx095
Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia
Erratum in
-
Erratum.Brain. 2018 Mar 1;141(3):e21. doi: 10.1093/brain/awx329. Brain. 2018. PMID: 29236964 Free PMC article. No abstract available.
Abstract
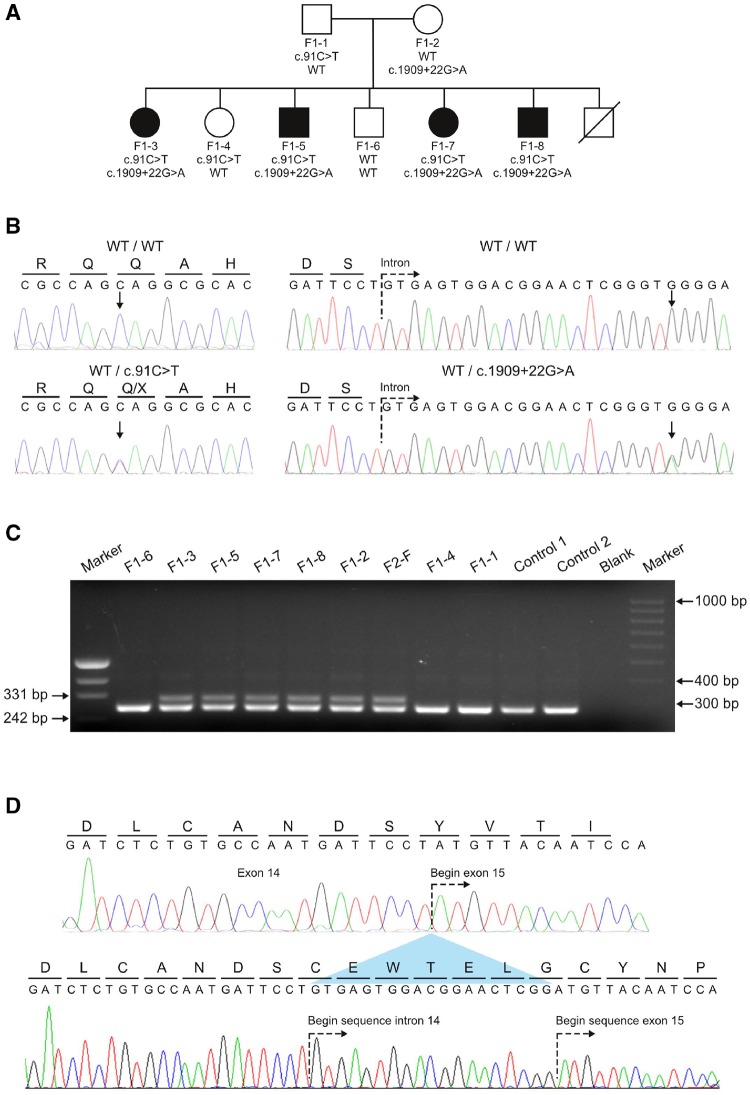
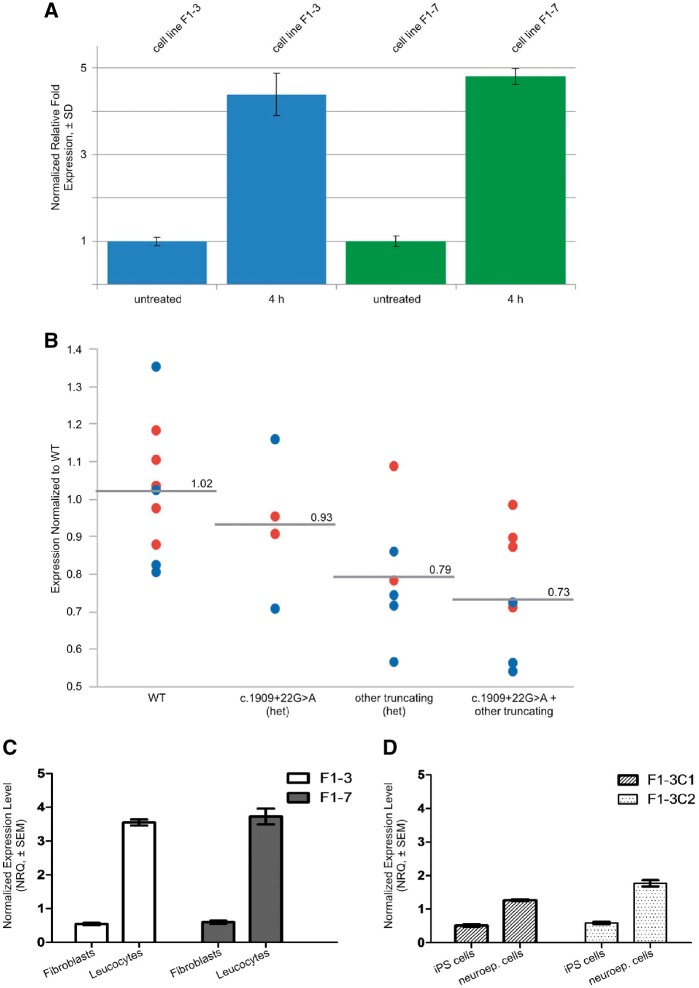
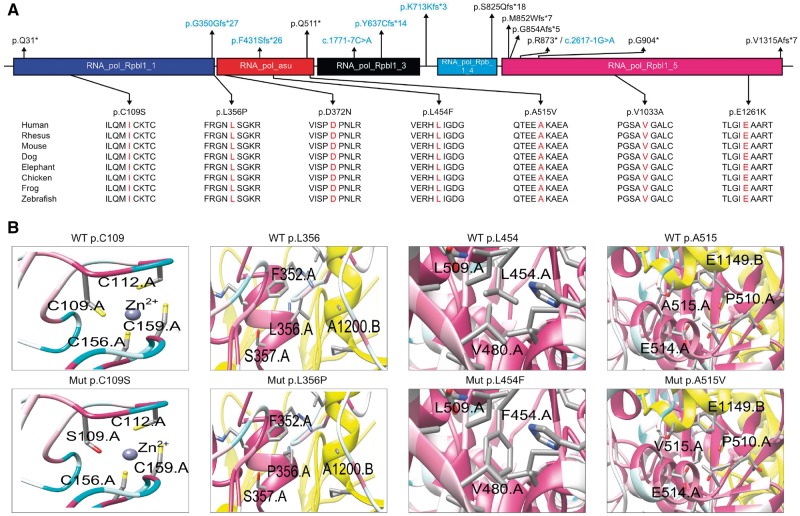
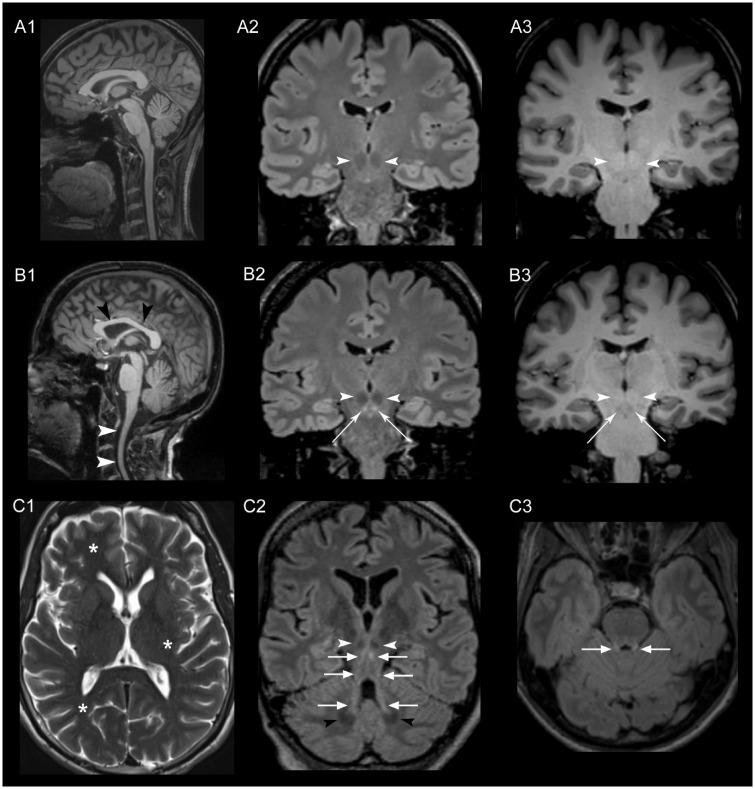
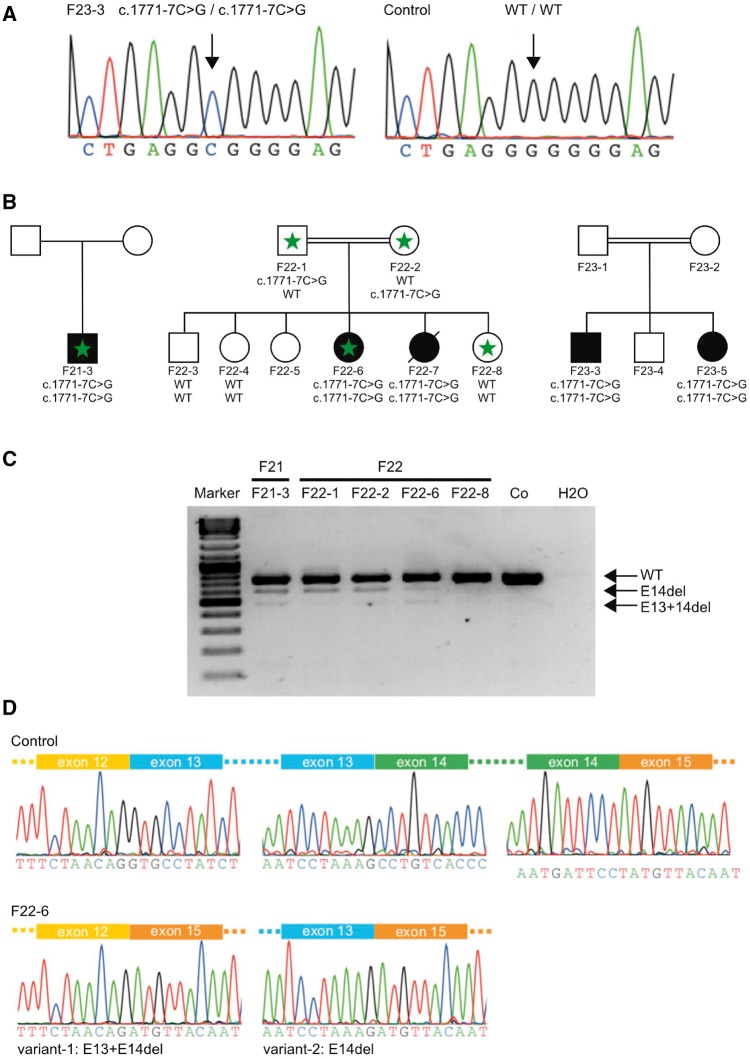
Despite extensive efforts, half of patients with rare movement disorders such as hereditary spastic paraplegias and cerebellar ataxias remain genetically unexplained, implicating novel genes and unrecognized mutations in known genes. Non-coding DNA variants are suspected to account for a substantial part of undiscovered causes of rare diseases. Here we identified mutations located deep in introns of POLR3A to be a frequent cause of hereditary spastic paraplegia and cerebellar ataxia. First, whole-exome sequencing findings in a recessive spastic ataxia family turned our attention to intronic variants in POLR3A, a gene previously associated with hypomyelinating leukodystrophy type 7. Next, we screened a cohort of hereditary spastic paraplegia and cerebellar ataxia cases (n = 618) for mutations in POLR3A and identified compound heterozygous POLR3A mutations in ∼3.1% of index cases. Interestingly, >80% of POLR3A mutation carriers presented the same deep-intronic mutation (c.1909+22G>A), which activates a cryptic splice site in a tissue and stage of development-specific manner and leads to a novel distinct and uniform phenotype. The phenotype is characterized by adolescent-onset progressive spastic ataxia with frequent occurrence of tremor, involvement of the central sensory tracts and dental problems (hypodontia, early onset of severe and aggressive periodontal disease). Instead of the typical hypomyelination magnetic resonance imaging pattern associated with classical POLR3A mutations, cases carrying c.1909+22G>A demonstrated hyperintensities along the superior cerebellar peduncles. These hyperintensities may represent the structural correlate to the cerebellar symptoms observed in these patients. The associated c.1909+22G>A variant was significantly enriched in 1139 cases with spastic ataxia-related phenotypes as compared to unrelated neurological and non-neurological phenotypes and healthy controls (P = 1.3 × 10-4). In this study we demonstrate that (i) autosomal-recessive mutations in POLR3A are a frequent cause of hereditary spastic ataxias, accounting for about 3% of hitherto genetically unclassified autosomal recessive and sporadic cases; and (ii) hypomyelination is frequently absent in POLR3A-related syndromes, especially when intronic mutations are present, and thus can no longer be considered as the unifying feature of POLR3A disease. Furthermore, our results demonstrate that substantial progress in revealing the causes of Mendelian diseases can be made by exploring the non-coding sequences of the human genome.
Keywords: POLR3A; cerebellar ataxia; hereditary spastic paraplegia; leukodystrophy; spastic ataxia.
© The Author (2017). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Comment in
-
POLR3A variants in hereditary spastic paraplegia and ataxia.Brain. 2018 Jan 1;141(1):e1. doi: 10.1093/brain/awx290. Brain. 2018. PMID: 29228109 Free PMC article. No abstract available.
-
Reply: POLR3A variants in hereditary spastic paraplegia and ataxia.Brain. 2018 Jan 1;141(1):e2. doi: 10.1093/brain/awx291. Brain. 2018. PMID: 29236946 Free PMC article. No abstract available.
-
Reply: Biallelic POLR3A variants confirmed as a frequent cause of hereditary ataxia and spastic paraparesis.Brain. 2019 Apr 1;142(4):e13. doi: 10.1093/brain/awz042. Brain. 2019. PMID: 30847463 No abstract available.
-
Biallelic POLR3A variants confirmed as a frequent cause of hereditary ataxia and spastic paraparesis.Brain. 2019 Apr 1;142(4):e12. doi: 10.1093/brain/awz041. Brain. 2019. PMID: 30847471 Free PMC article. No abstract available.
References
-
- Azmanov DN, Siira SJ, Chamova T, Kaprelyan A, Guergueltcheva V, Shearwood AJ. et al. Transcriptome-wide effects of a POLR3A gene mutation in patients with an unusual phenotype of striatal involvement. Hum Mol Genet 2016; 25: 4302–14. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
