

Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper
- PMID: 28460117
- PMCID: PMC5850834
- DOI: 10.1093/molbev/msx148
Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper
Abstract
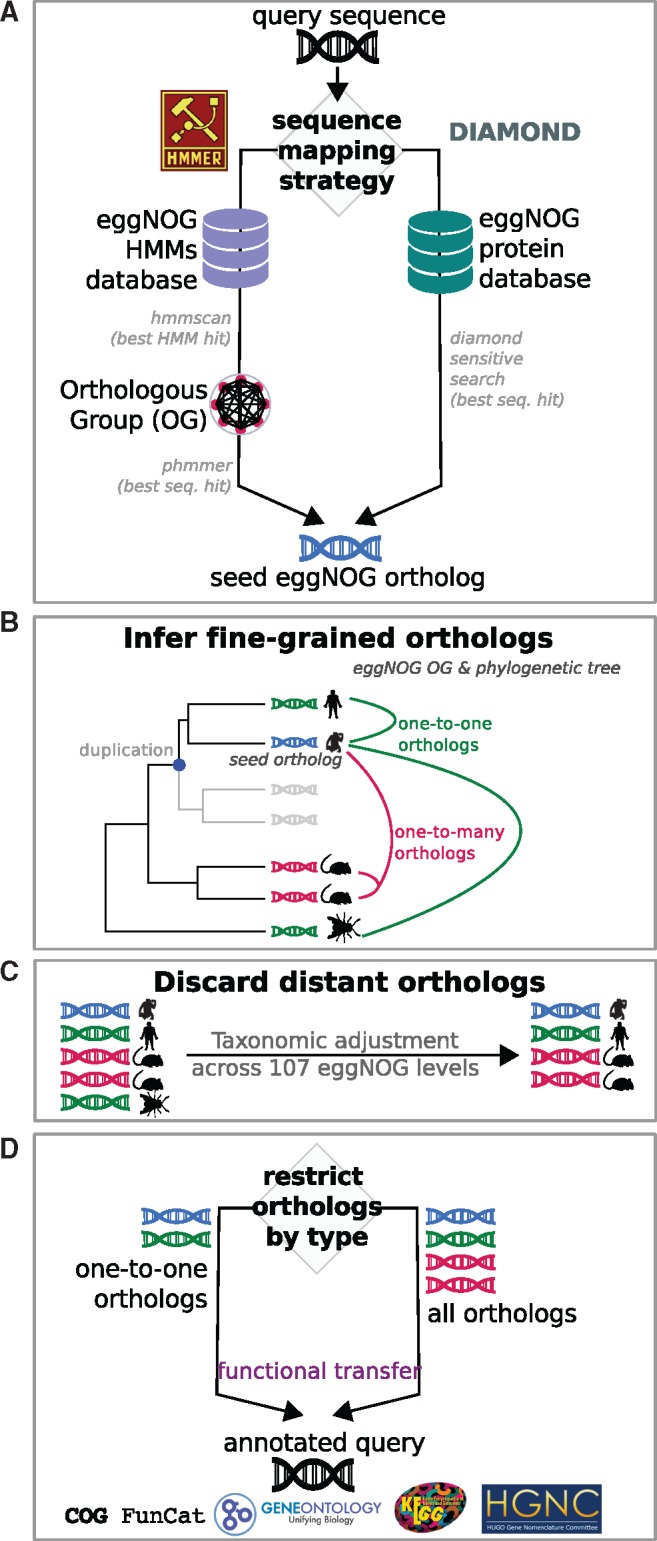
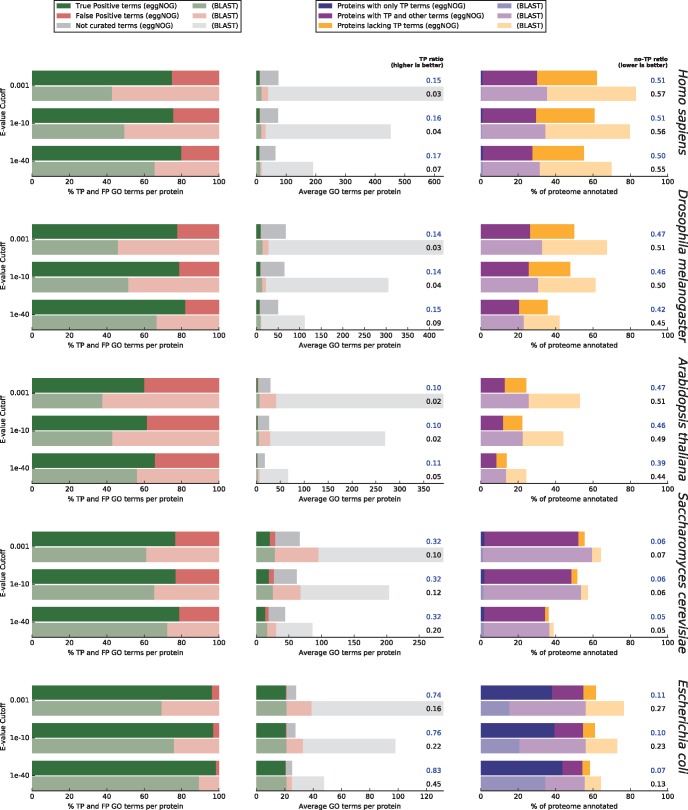
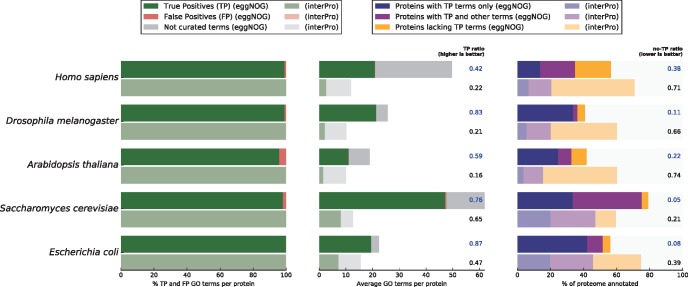
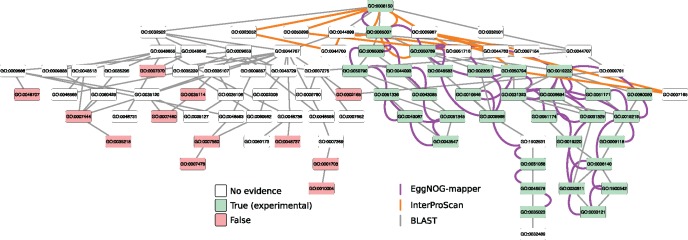
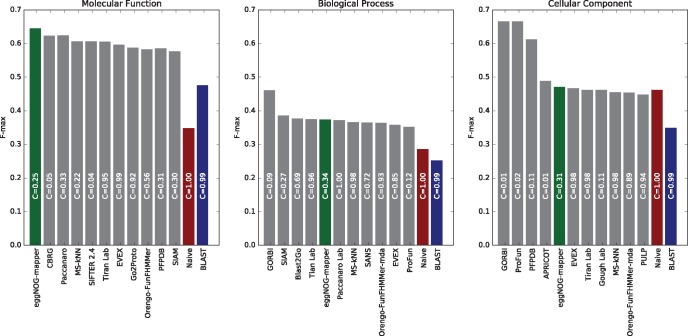
Orthology assignment is ideally suited for functional inference. However, because predicting orthology is computationally intensive at large scale, and most pipelines are relatively inaccessible (e.g., new assignments only available through database updates), less precise homology-based functional transfer is still the default for (meta-)genome annotation. We, therefore, developed eggNOG-mapper, a tool for functional annotation of large sets of sequences based on fast orthology assignments using precomputed clusters and phylogenies from the eggNOG database. To validate our method, we benchmarked Gene Ontology (GO) predictions against two widely used homology-based approaches: BLAST and InterProScan. Orthology filters applied to BLAST results reduced the rate of false positive assignments by 11%, and increased the ratio of experimentally validated terms recovered over all terms assigned per protein by 15%. Compared with InterProScan, eggNOG-mapper achieved similar proteome coverage and precision while predicting, on average, 41 more terms per protein and increasing the rate of experimentally validated terms recovered over total term assignments per protein by 35%. EggNOG-mapper predictions scored within the top-5 methods in the three GO categories using the CAFA2 NK-partial benchmark. Finally, we evaluated eggNOG-mapper for functional annotation of metagenomics data, yielding better performance than interProScan. eggNOG-mapper runs ∼15× faster than BLAST and at least 2.5× faster than InterProScan. The tool is available standalone and as an online service at http://eggnog-mapper.embl.de.
Keywords: comparative genomics; functional annotation; gene function; genomics; metagenomics; orthology.
© The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Buchfink B, Xie C, Huson DH.. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12:59–60. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
