

A Robust Framework for Microbial Archaeology
- PMID: 28460196
- PMCID: PMC5581243
- DOI: 10.1146/annurev-genom-091416-035526
A Robust Framework for Microbial Archaeology
Abstract
Microbial archaeology is flourishing in the era of high-throughput sequencing, revealing the agents behind devastating historical plagues, identifying the cryptic movements of pathogens in prehistory, and reconstructing the ancestral microbiota of humans. Here, we introduce the fundamental concepts and theoretical framework of the discipline, then discuss applied methodologies for pathogen identification and microbiome characterization from archaeological samples. We give special attention to the process of identifying, validating, and authenticating ancient microbes using high-throughput DNA sequencing data. Finally, we outline standards and precautions to guide future research in the field.
Keywords: ancient DNA; bacteria; high-throughput sequencing; metagenomics; microbiology; microbiome; pathogens.
Figures
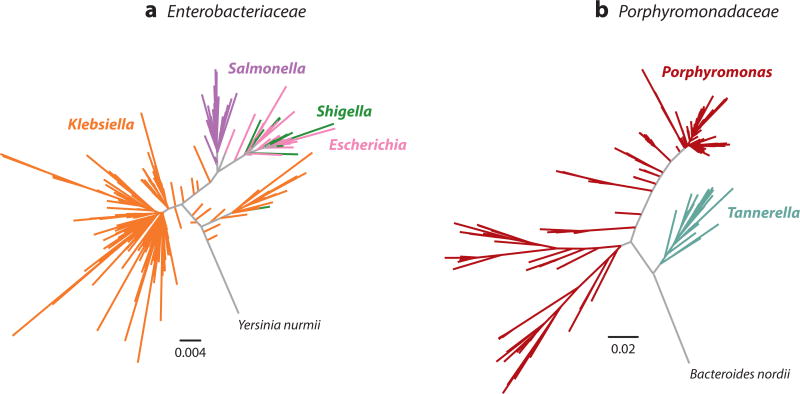
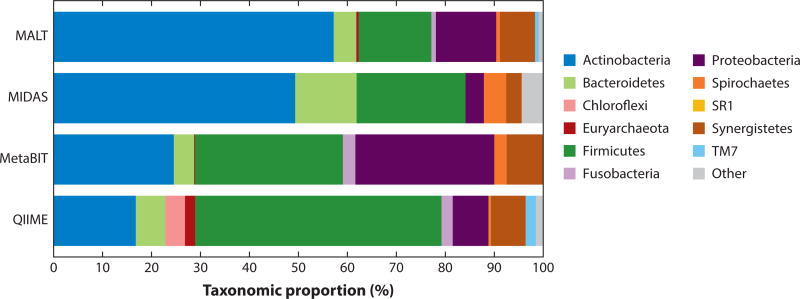
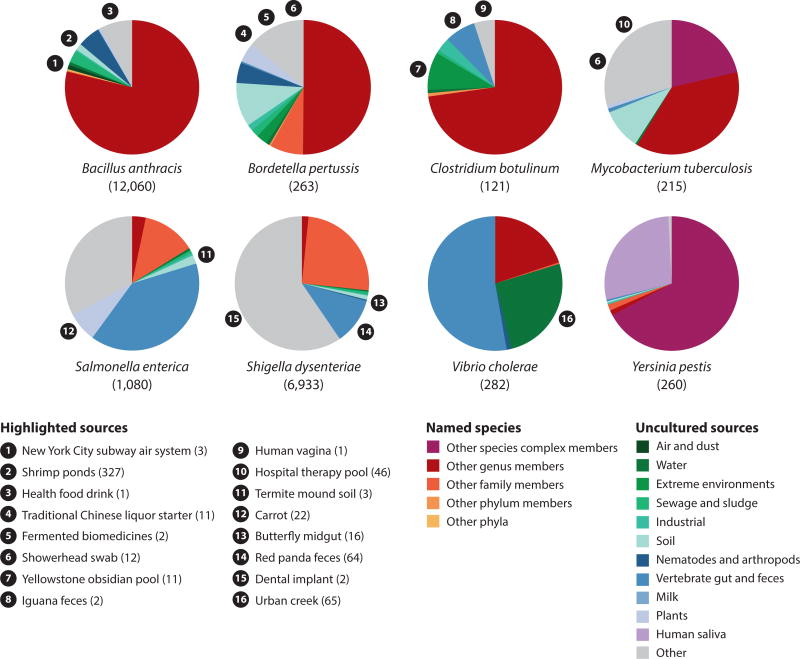
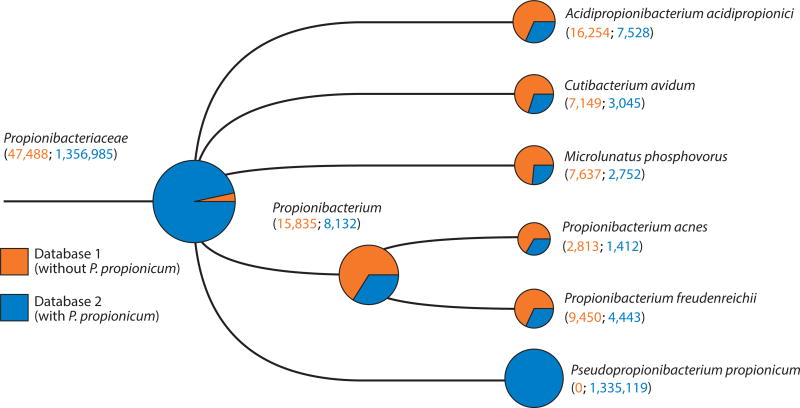
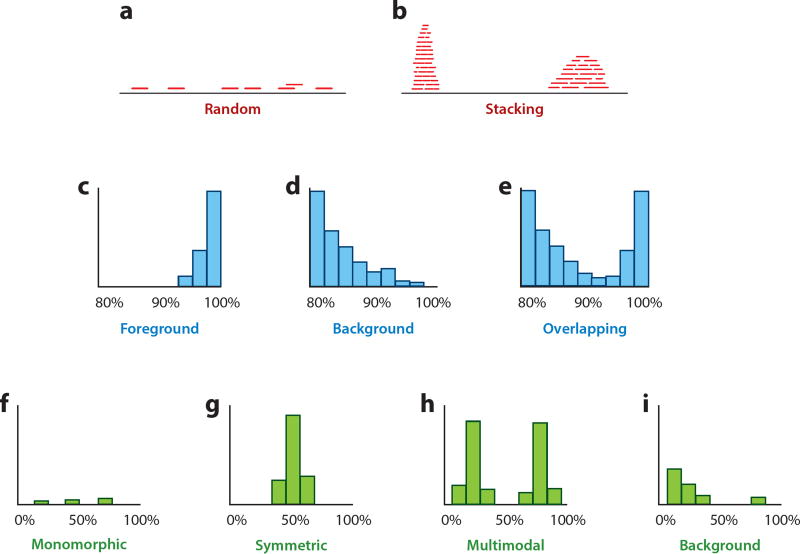
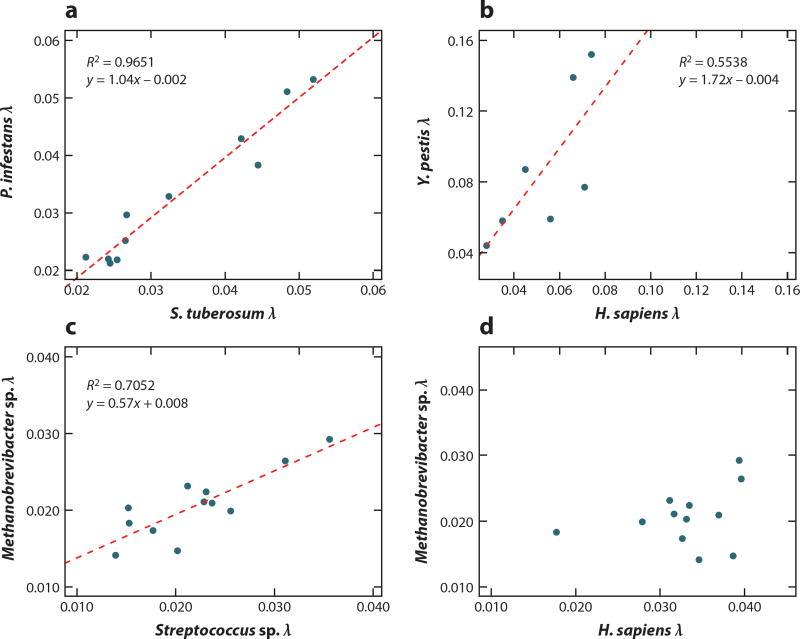
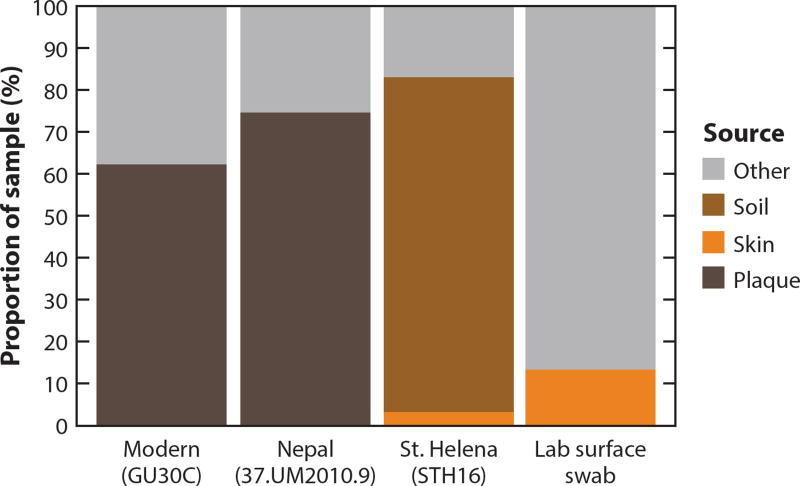
References
-
- Achtman M. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. Microbiol. 2008;66 62:53–70. - PubMed
-
- Achtman M, Wagner M. Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 2008;6:431–40. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
