

Differential manipulation of arrestin-3 binding to basal and agonist-activated G protein-coupled receptors
- PMID: 28461104
- PMCID: PMC5797668
- DOI: 10.1016/j.cellsig.2017.04.021
Differential manipulation of arrestin-3 binding to basal and agonist-activated G protein-coupled receptors
Abstract
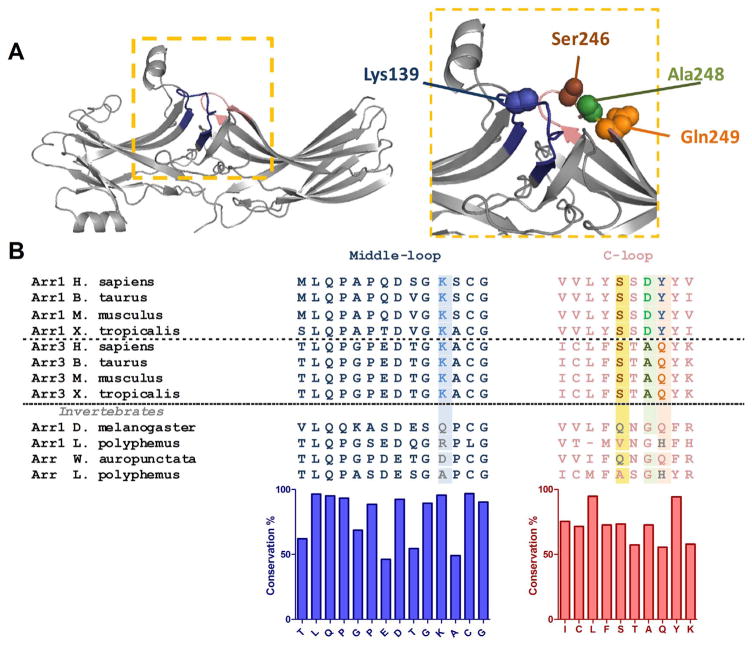
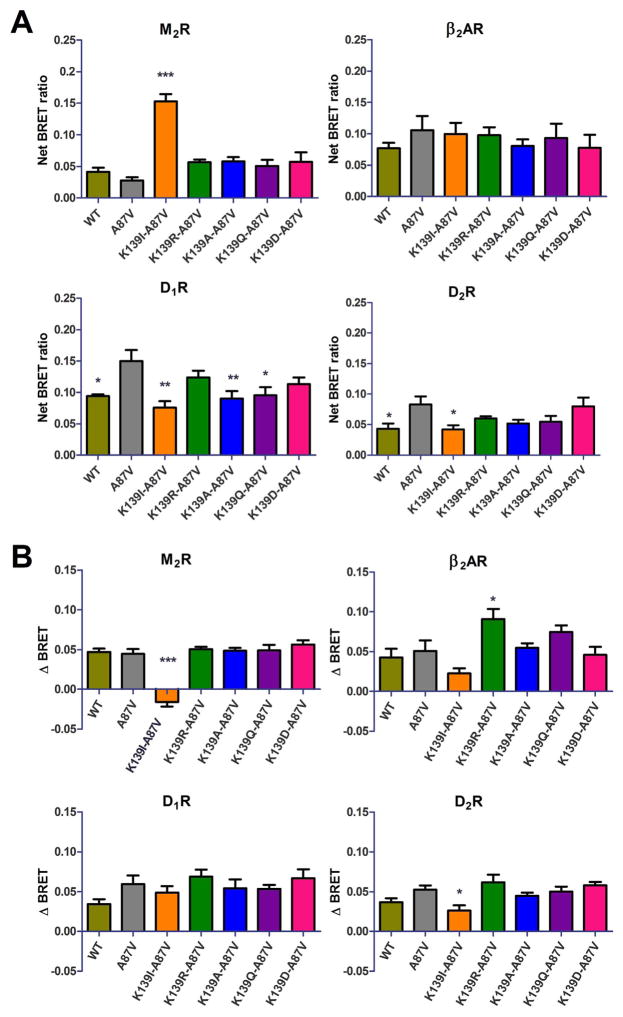
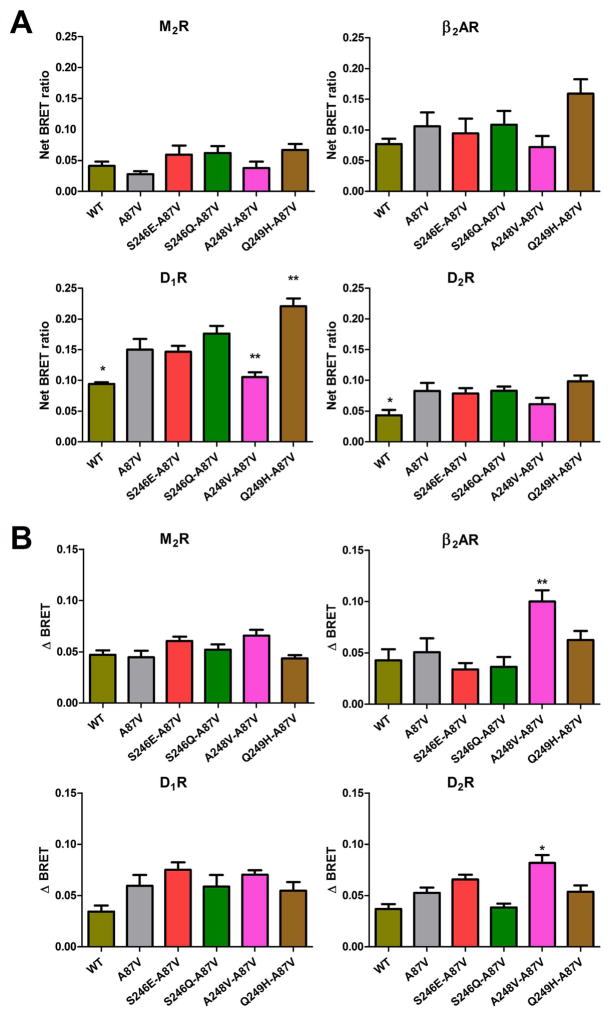
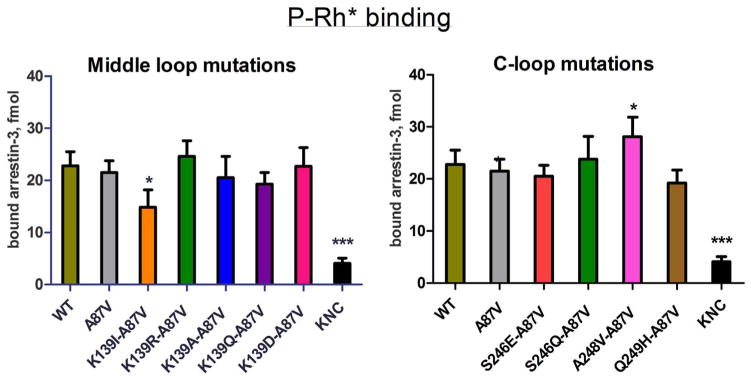
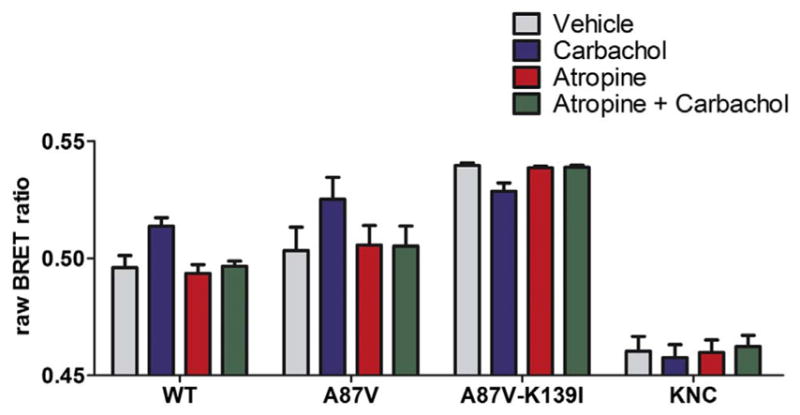
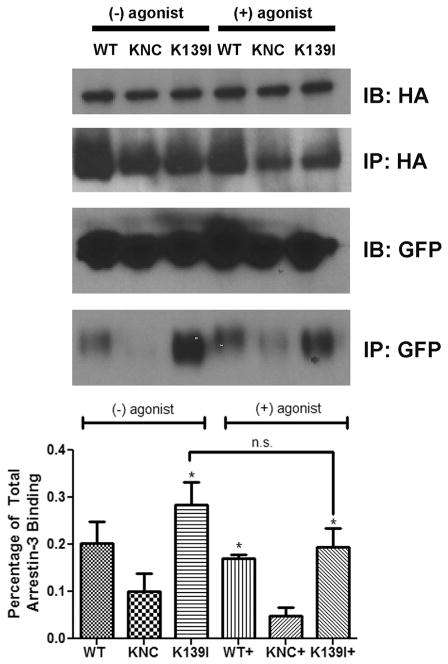
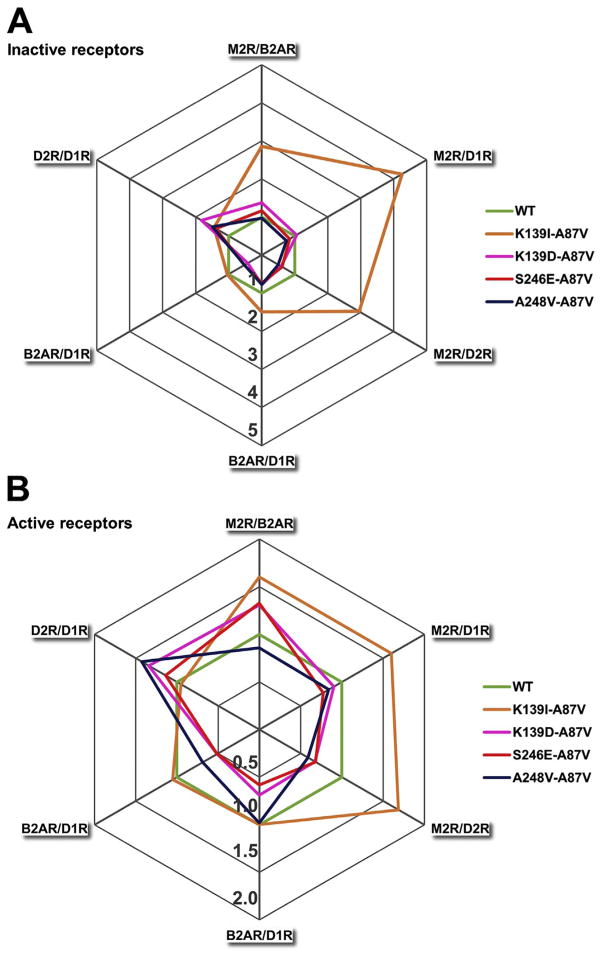
Non-visual arrestins interact with hundreds of different G protein-coupled receptors (GPCRs). Here we show that by introducing mutations into elements that directly bind receptors, the specificity of arrestin-3 can be altered. Several mutations in the two parts of the central "crest" of the arrestin molecule, middle-loop and C-loop, enhanced or reduced arrestin-3 interactions with several GPCRs in receptor subtype and functional state-specific manner. For example, the Lys139Ile substitution in the middle-loop dramatically enhanced the binding to inactive M2 muscarinic receptor, so that agonist activation of the M2 did not further increase arrestin-3 binding. Thus, the Lys139Ile mutation made arrestin-3 essentially an activation-independent binding partner of M2, whereas its interactions with other receptors, including the β2-adrenergic receptor and the D1 and D2 dopamine receptors, retained normal activation dependence. In contrast, the Ala248Val mutation enhanced agonist-induced arrestin-3 binding to the β2-adrenergic and D2 dopamine receptors, while reducing its interaction with the D1 dopamine receptor. These mutations represent the first example of altering arrestin specificity via enhancement of the arrestin-receptor interactions rather than selective reduction of the binding to certain subtypes.
Keywords: Arrestin; GPCRs; Protein engineering; Protein-protein interactions; Receptor specificity.
Copyright © 2017. Published by Elsevier Inc.
Figures
References
-
- Schoneberg T, Schulz A, Biebermann H, Hermsdorf T, Rompler H, Sangkuhl K. Pharmacol Ther. 2004;104:173–206. - PubMed
-
- Russo D, Arturi F, Schlumberger M, Caillou B, Monier R, Filetti S, Suarez HG. Oncogene. 1995;11:1907–1911. - PubMed
-
- Hebrant A, van Staveren WC, Maenhaut C, Dumont JE, Leclere J. Eur J Endocrinol. 2011;164:1–9. - PubMed
-
- Carman CV, Benovic JL. Curr Opin Neurobiol. 1998;8:335–344. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
