

Transcriptome analysis of smut fungi reveals widespread intergenic transcription and conserved antisense transcript expression
- PMID: 28464849
- PMCID: PMC5414199
- DOI: 10.1186/s12864-017-3720-8
Transcriptome analysis of smut fungi reveals widespread intergenic transcription and conserved antisense transcript expression
Abstract
Background: Biotrophic fungal plant pathogens cause billions of dollars in losses to North American crops annually. The model for functional investigation of these fungi is Ustilago maydis. Its 20.5 Mb annotated genome sequence has been an excellent resource for investigating biotrophic plant pathogenesis. Expressed-sequence tag libraries and microarray hybridizations have provided insight regarding the type of transcripts produced by U. maydis but these analyses were not comprehensive and there were insufficient data for transcriptome comparison to other smut fungi. To improve transcriptome annotation and enable comparative analyses, comprehensive strand-specific RNA-seq was performed on cell-types of three related smut species: U. maydis (common smut of corn), Ustilago hordei (covered smut of barley), and Sporisorium reilianum (head smut of corn).
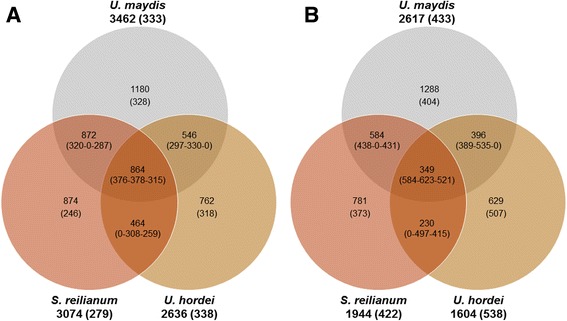
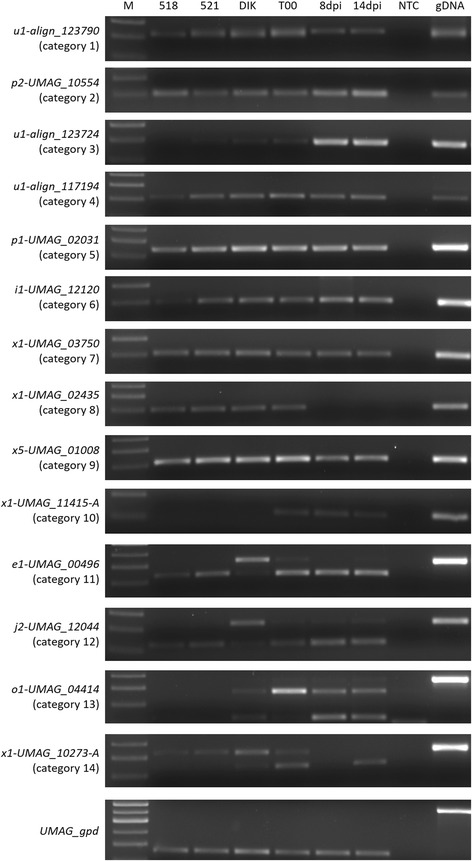
Results: In total, >1 billion paired-end sequence reads were obtained from haploid cell, dikaryon and teliospore RNA of U. maydis, haploid cell RNA of U. hordei, and haploid and dikaryon cell RNA of S. reilianum. The sequences were assembled into transfrags using Trinity, and updated gene models were created using PASA and categorized with Cufflinks Cuffcompare. Representative genes that were predicted for the first time with these RNA-seq analyses and genes with novel annotation features were independently assessed by reverse transcriptase PCR. The analyses indicate hundreds more predicted proteins, relative to the previous genome annotation, could be produced by U. maydis from altered transcript forms, and that the number of non-coding RNAs produced, including transcribed intergenic sequences and natural antisense transcripts, approximately equals the number of mRNAs. This high representation of non-coding RNAs appears to be a conserved feature of the smut fungi regardless of whether they have RNA interference machinery. Approximately 50% of the identified NATs were conserved among the smut fungi.
Conclusions: Overall, these analyses revealed: 1) smut genomes encode a number of transcriptional units that is twice the number of annotated protein-coding genes, 2) a small number of intergenic transcripts may encode proteins with characteristics of fungal effectors, 3) the vast majority of intergenic and antisense transcripts do not contain ORFs, 4) a large proportion of the identified antisense transcripts were detected at orthologous loci among the smut fungi, and 5) there is an enrichment of functional categories among orthologous loci that suggests antisense RNAs could have a genome-wide, non-RNAi-mediated, influence on gene expression in smut fungi.
Keywords: Natural antisense transcripts; Non-coding RNAs; RNA-seq; Smut fungi; Sporisorium reilianum; Ustilago hordei; Ustilago maydis.
Figures
Similar articles
-
Gene discovery and transcript analyses in the corn smut pathogen Ustilago maydis: expressed sequence tag and genome sequence comparison.BMC Genomics. 2007 Sep 24;8:334. doi: 10.1186/1471-2164-8-334. BMC Genomics. 2007. PMID: 17892571 Free PMC article.
-
Exploring links between antisense RNAs and pathogenesis in Ustilago maydis through transcript and gene characterization.Fungal Genet Biol. 2020 Jan;134:103283. doi: 10.1016/j.fgb.2019.103283. Epub 2019 Oct 16. Fungal Genet Biol. 2020. PMID: 31629082
-
Genome comparison of barley and maize smut fungi reveals targeted loss of RNA silencing components and species-specific presence of transposable elements.Plant Cell. 2012 May;24(5):1733-45. doi: 10.1105/tpc.112.097261. Epub 2012 May 22. Plant Cell. 2012. PMID: 22623492 Free PMC article.
-
Molecular Interactions Between Smut Fungi and Their Host Plants.Annu Rev Phytopathol. 2019 Aug 25;57:411-430. doi: 10.1146/annurev-phyto-082718-100139. Epub 2019 Jul 23. Annu Rev Phytopathol. 2019. PMID: 31337276 Review.
-
The posttranscriptional machinery of Ustilago maydis.Fungal Genet Biol. 2008 Aug;45 Suppl 1:S40-6. doi: 10.1016/j.fgb.2008.03.013. Epub 2008 Mar 31. Fungal Genet Biol. 2008. PMID: 18468465 Review.
Cited by
-
Telomerase RNA plays a major role in the completion of the life cycle in Ustilago maydis and shares conserved domains with other Ustilaginales.PLoS One. 2023 Mar 23;18(3):e0281251. doi: 10.1371/journal.pone.0281251. eCollection 2023. PLoS One. 2023. PMID: 36952474 Free PMC article.
-
Alternative splicing diversifies the transcriptome and proteome of the rice blast fungus during host infection.RNA Biol. 2022;19(1):373-385. doi: 10.1080/15476286.2022.2043040. Epub 2021 Dec 31. RNA Biol. 2022. PMID: 35311472 Free PMC article.
-
RNA Sequencing Reveals Widespread Transcription of Natural Antisense RNAs in Entamoeba Species.Microorganisms. 2022 Feb 8;10(2):396. doi: 10.3390/microorganisms10020396. Microorganisms. 2022. PMID: 35208849 Free PMC article.
-
GC-MS-Based Metabolomics for the Smut Fungus Ustilago maydis: A Comprehensive Method Optimization to Quantify Intracellular Metabolites.Front Mol Biosci. 2020 Aug 19;7:211. doi: 10.3389/fmolb.2020.00211. eCollection 2020. Front Mol Biosci. 2020. PMID: 32974387 Free PMC article.
-
Characterization of RNA Helicase Genes in Ustilago maydis Reveals Links to Stress Response and Teliospore Dormancy.Int J Mol Sci. 2025 Mar 8;26(6):2432. doi: 10.3390/ijms26062432. Int J Mol Sci. 2025. PMID: 40141077 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
