

Plasma Membrane Association by N-Acylation Governs PKG Function in Toxoplasma gondii
- PMID: 28465425
- PMCID: PMC5414004
- DOI: 10.1128/mBio.00375-17
Plasma Membrane Association by N-Acylation Governs PKG Function in Toxoplasma gondii
Abstract
Cyclic GMP (cGMP)-dependent protein kinase (protein kinase G [PKG]) is essential for microneme secretion, motility, invasion, and egress in apicomplexan parasites, However, the separate roles of two isoforms of the kinase that are expressed by some apicomplexans remain uncertain. Despite having identical regulatory and catalytic domains, PKGI is plasma membrane associated whereas PKGII is cytosolic in Toxoplasma gondii To determine whether these isoforms are functionally distinct or redundant, we developed an auxin-inducible degron (AID) tagging system for conditional protein depletion in T. gondii By combining AID regulation with genome editing strategies, we determined that PKGI is necessary and fully sufficient for PKG-dependent cellular processes. Conversely, PKGII is functionally insufficient and dispensable in the presence of PKGI The difference in functionality mapped to the first 15 residues of PKGI, containing a myristoylated Gly residue at position 2 that is critical for membrane association and PKG function. Collectively, we have identified a novel requirement for cGMP signaling at the plasma membrane and developed a new system for examining essential proteins in T. gondiiIMPORTANCEToxoplasma gondii is an obligate intracellular apicomplexan parasite and important clinical and veterinary pathogen that causes toxoplasmosis. Since apicomplexans can only propagate within host cells, efficient invasion is critically important for their life cycles. Previous studies using chemical genetics demonstrated that cyclic GMP signaling through protein kinase G (PKG)-controlled invasion by apicomplexan parasites. However, these studies did not resolve functional differences between two compartmentalized isoforms of the kinase. Here we developed a conditional protein regulation tool to interrogate PKG isoforms in T. gondii We found that the cytosolic PKG isoform was largely insufficient and dispensable. In contrast, the plasma membrane-associated isoform was necessary and fully sufficient for PKG function. Our studies identify the plasma membrane as a key location for PKG activity and provide a broadly applicable system for examining essential proteins in T. gondii.
Copyright © 2017 Brown et al.
Figures
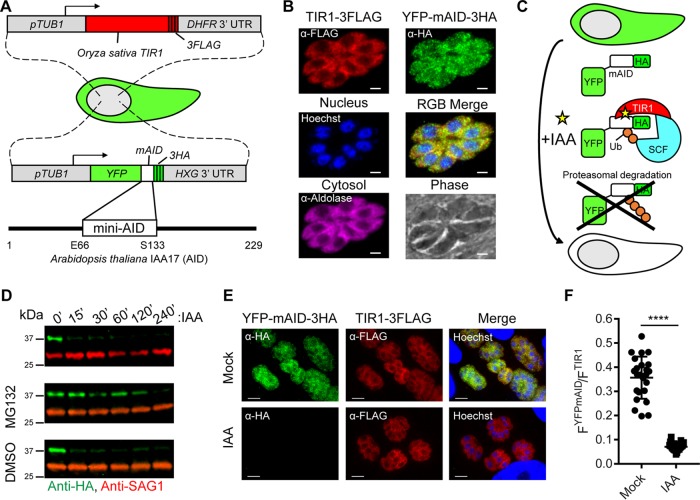
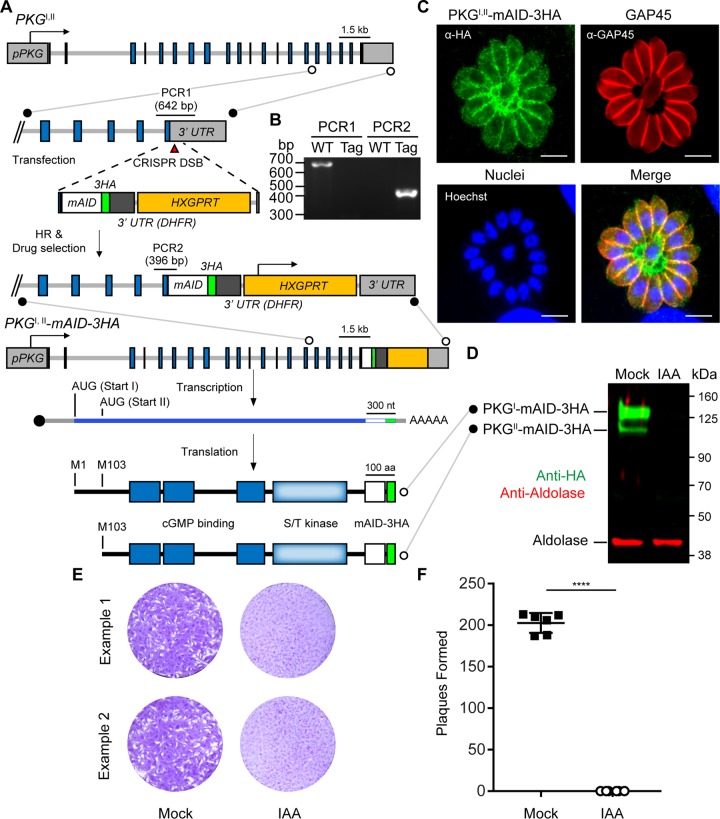
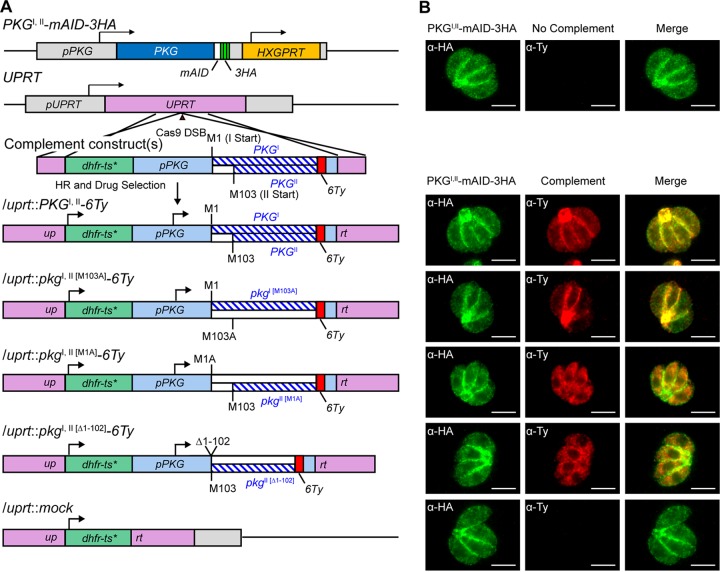
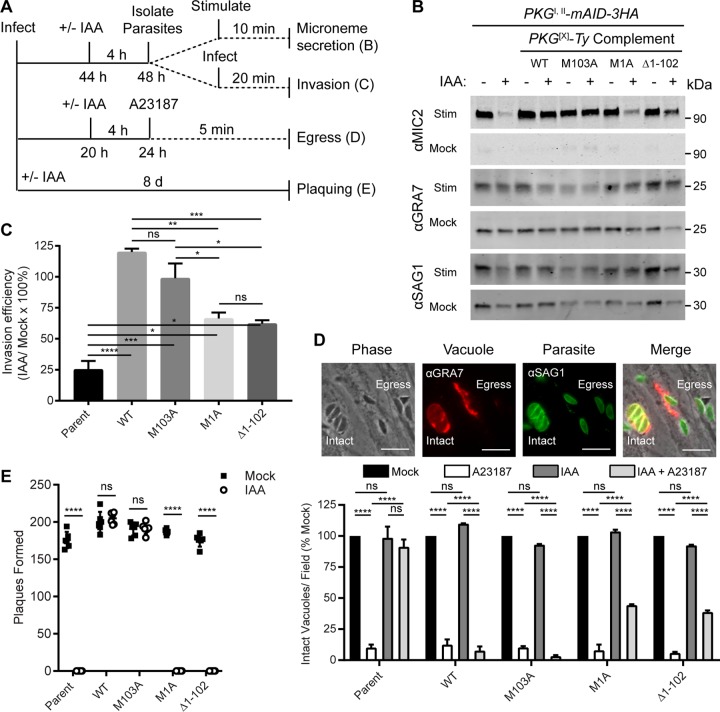
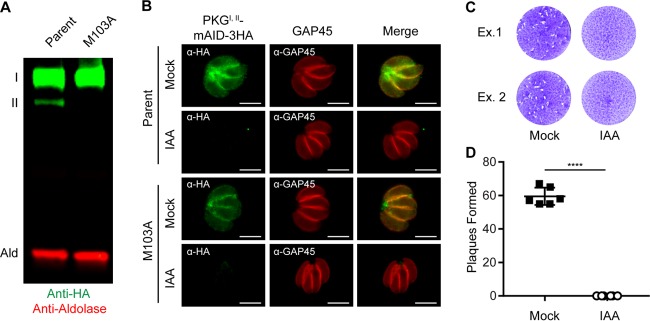
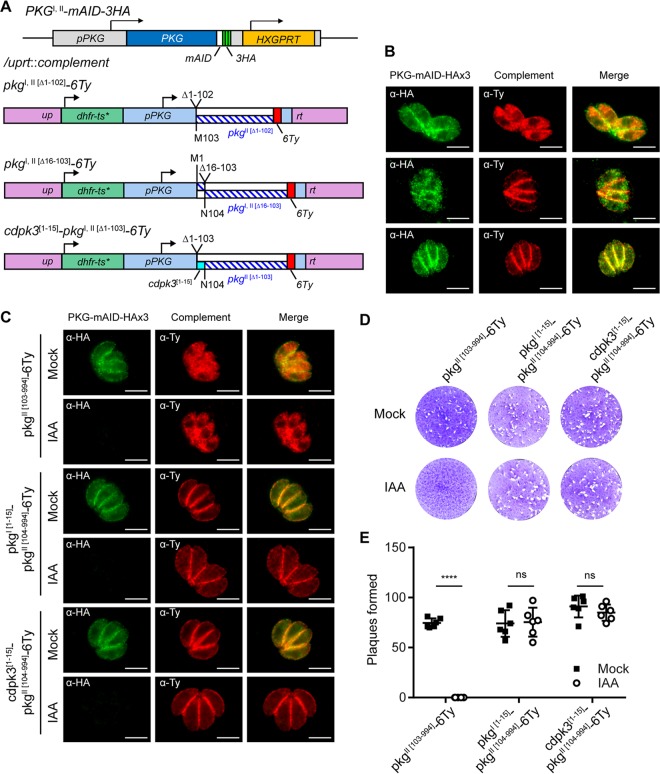
References
-
- Jacot D, Tosetti N, Pires I, Stock J, Graindorge A, Hung YF, Han H, Tewari R, Kursula I, Soldati-Favre D. 2016. An apicomplexan actin-binding protein serves as a connector and lipid sensor to coordinate motility and invasion. Cell Host Microbe 20:731–743. doi: 10.1016/j.chom.2016.10.020. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
