

2016 WHO Clinical Molecular and Pathological Criteria for Classification and Staging of Myeloproliferative Neoplasms (MPN) Caused by MPN Driver Mutations in the JAK2, MPL and CALR Genes in the Context of New 2016 WHO Classification: Prognostic and Therapeutic Implications
- PMID: 28465746
- PMCID: PMC5394501
2016 WHO Clinical Molecular and Pathological Criteria for Classification and Staging of Myeloproliferative Neoplasms (MPN) Caused by MPN Driver Mutations in the JAK2, MPL and CALR Genes in the Context of New 2016 WHO Classification: Prognostic and Therapeutic Implications
Abstract
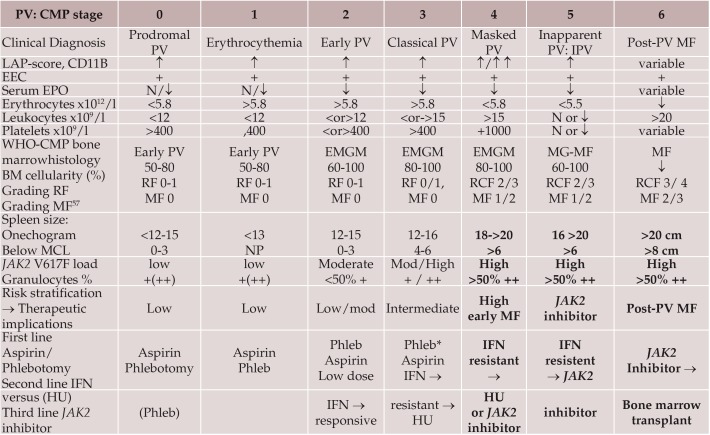
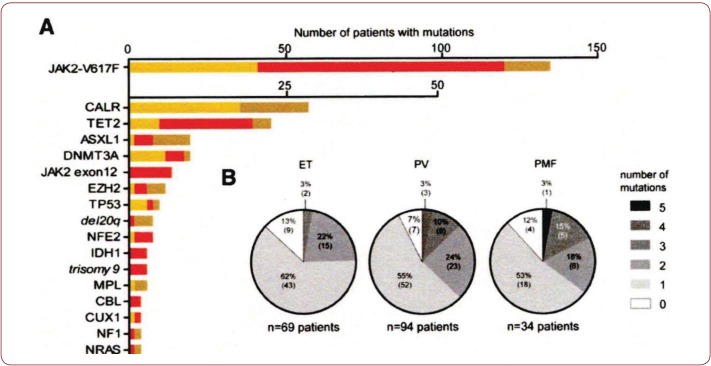
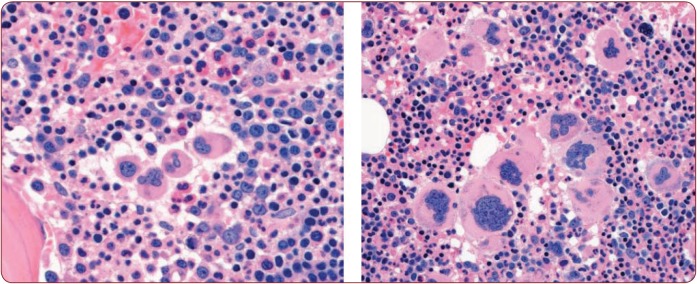
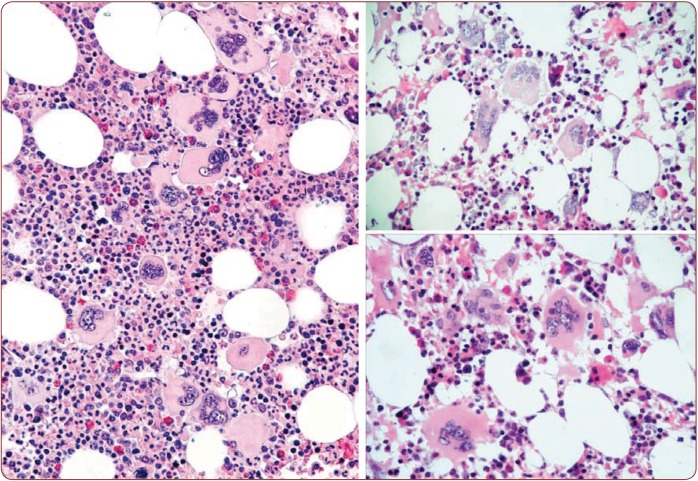
The 2016 WHO-CMP classification proposal defines a broad spectrum of JAK2 V617F mutated MPN phenotypes: normocellular ET, hypercellular ET due to increased erythropoiesis (prodromal PV), hypercellular ET with megakaryocytic-granulocytic myeloproliferation and splenomegaly (EMGM or masked PV), erythrocythemic PV, early and overt classical PV, advanced PV with MF and post-PV MF. ET heterozygous for the JAK2 V617F mutation is associated with low JAK2 mutation load and normal life expectance. PV patients are hetero-homozygous versus homozygous for the JAK2 V617F mutation in their early versus advanced stages with increasing JAK2 mutation load from less than 50% to 100% and increase of MPN disease burden during life long follow-up in terms of symptomatic splenomegaly, constitutional symptoms, bone marrow hypercellularity and secondary MF. Pretreatment bone marrow biopsy in prefibrotic MPNs is of diagnostic and prognostic importance. JAK2 exon 12 mutated MPN is a distinct benign early stage PV. CALR mutated hypercellular thrombocythemia show distinct PMGM bone marrow characteristics of clustered larged immature dysmorphic megakaryocytes with bulky (bulbous) hyperchromatic nuclei, which are not seen in JAK2 mutated ET and PV. MPL mutated normocellular thrombocythemia is featured by clustered giant megakaryocytes with hyperlobulated stag-horn-like nuclei without features of PV in blood and bone marrow. Myeloproliferative disease burden in each of the JAK2, CALR and MPL MPNs is best reflected by the degree of anemia, splenomegaly, mutation allele burden, bone marrow cellularity and myelofibrosis.
Figures

















Similar articles
-
Changing concepts of diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms: from Dameshek 1950 to Vainchenker 2005 and beyond.Acta Haematol. 2015;133(1):36-51. doi: 10.1159/000358580. Epub 2014 Aug 7. Acta Haematol. 2015. PMID: 25116092 Review.
-
Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thrombocythemia, polycythemia vera and essential megakaryocytic granulocytic metaplasia and myelofibrosis.Neth J Med. 1999 Feb;54(2):46-62. doi: 10.1016/s0300-2977(98)00143-0. Neth J Med. 1999. PMID: 10079679 Review.
-
WHO bone marrow features and European clinical, molecular, and pathological (ECMP) criteria for the diagnosis of myeloproliferative disorders.Leuk Res. 2007 Aug;31(8):1031-8. doi: 10.1016/j.leukres.2007.01.021. Epub 2007 Mar 23. Leuk Res. 2007. PMID: 17367853 Review.
-
Characterization and Prognosis Significance of JAK2 (V617F), MPL, and CALR Mutations in Philadelphia-Negative Myeloproliferative Neoplasms.Asian Pac J Cancer Prev. 2016 Oct 1;17(10):4647-4653. doi: 10.22034/apjcp.2016.17.10.4647. Asian Pac J Cancer Prev. 2016. PMID: 27892678 Free PMC article.
-
The 2001 World Health Organization and updated European clinical and pathological criteria for the diagnosis, classification, and staging of the Philadelphia chromosome-negative chronic myeloproliferative disorders.Semin Thromb Hemost. 2006 Jun;32(4 Pt 2):307-40. doi: 10.1055/s-2006-942754. Semin Thromb Hemost. 2006. PMID: 16810609 Review.
Cited by
-
Emerging therapeutic targets in myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas.Expert Opin Ther Targets. 2018 Jan;22(1):45-57. doi: 10.1080/14728222.2018.1406924. Epub 2017 Nov 24. Expert Opin Ther Targets. 2018. PMID: 29148847 Free PMC article. Review.
References
-
- Dameshek W, Henstell HH - The diagnosis of polycythemia. Ann Intern Med. 1940;13:1360–1387.
-
- Dameshek W - Physiopathology and corse of polycythemia vera as related to therapy. JAMA. 1950;142:790–797. - PubMed
-
- Dameshek W - The treatment of Polycythemia. Blood. 1946;1:256.
-
- Dameshek W - Some speculations on the myeloproliferative syndromes. Blood. 1951;6:372–375. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous