

Inferring the physical properties of yeast chromatin through Bayesian analysis of whole nucleus simulations
- PMID: 28468672
- PMCID: PMC5414205
- DOI: 10.1186/s13059-017-1199-x
Inferring the physical properties of yeast chromatin through Bayesian analysis of whole nucleus simulations
Abstract
Background: The structure and mechanical properties of chromatin impact DNA functions and nuclear architecture but remain poorly understood. In budding yeast, a simple polymer model with minimal sequence-specific constraints and a small number of structural parameters can explain diverse experimental data on nuclear architecture. However, how assumed chromatin properties affect model predictions was not previously systematically investigated.
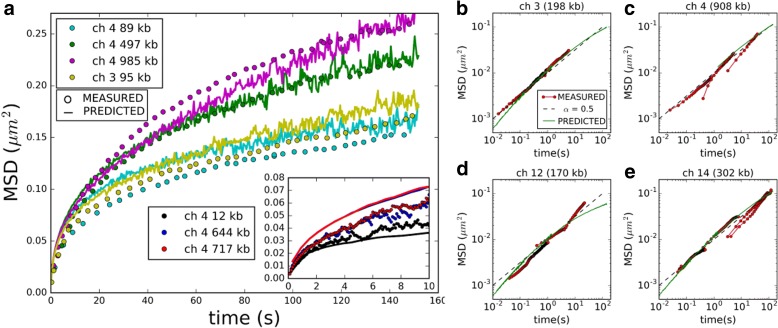
Results: We used hundreds of dynamic chromosome simulations and Bayesian inference to determine chromatin properties consistent with an extensive dataset that includes hundreds of measurements from imaging in fixed and live cells and two Hi-C studies. We place new constraints on average chromatin fiber properties, narrowing down the chromatin compaction to ~53-65 bp/nm and persistence length to ~52-85 nm. These constraints argue against a 20-30 nm fiber as the exclusive chromatin structure in the genome. Our best model provides a much better match to experimental measurements of nuclear architecture and also recapitulates chromatin dynamics measured on multiple loci over long timescales.
Conclusion: This work substantially improves our understanding of yeast chromatin mechanics and chromosome architecture and provides a new analytic framework to infer chromosome properties in other organisms.
Keywords: Chromatin; Chromosomes; Nuclear architecture; Polymer models; Yeast.
Figures




Similar articles
-
Nuclear organization and chromatin dynamics in yeast: biophysical models or biologically driven interactions?Biochim Biophys Acta. 2012 Jun;1819(6):468-81. doi: 10.1016/j.bbagrm.2011.12.010. Epub 2012 Jan 5. Biochim Biophys Acta. 2012. PMID: 22245105 Review.
-
A predictive computational model of the dynamic 3D interphase yeast nucleus.Curr Biol. 2012 Oct 23;22(20):1881-90. doi: 10.1016/j.cub.2012.07.069. Epub 2012 Aug 30. Curr Biol. 2012. PMID: 22940469
-
From dynamic chromatin architecture to DNA damage repair and back.Nucleus. 2018 Jan 1;9(1):161-170. doi: 10.1080/19491034.2017.1419847. Nucleus. 2018. PMID: 29271297 Free PMC article. Review.
-
Long-range compaction and flexibility of interphase chromatin in budding yeast analyzed by high-resolution imaging techniques.Proc Natl Acad Sci U S A. 2004 Nov 23;101(47):16495-500. doi: 10.1073/pnas.0402766101. Epub 2004 Nov 15. Proc Natl Acad Sci U S A. 2004. PMID: 15545610 Free PMC article.
-
How to build a yeast nucleus.Nucleus. 2013 Sep-Oct;4(5):361-6. doi: 10.4161/nucl.26226. Epub 2013 Aug 22. Nucleus. 2013. PMID: 23974728 Free PMC article.
Cited by
-
Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging.Science. 2022 Apr 29;376(6592):496-501. doi: 10.1126/science.abn6583. Epub 2022 Apr 14. Science. 2022. PMID: 35420890 Free PMC article.
-
Chromatin mobility upon DNA damage: state of the art and remaining questions.Curr Genet. 2019 Feb;65(1):1-9. doi: 10.1007/s00294-018-0852-6. Epub 2018 Jun 8. Curr Genet. 2019. PMID: 29947969 Review.
-
DNA Repair: The Search for Homology.Bioessays. 2018 May;40(5):e1700229. doi: 10.1002/bies.201700229. Epub 2018 Mar 30. Bioessays. 2018. PMID: 29603285 Free PMC article. Review.
-
Rouse model with transient intramolecular contacts on a timescale of seconds recapitulates folding and fluctuation of yeast chromosomes.Nucleic Acids Res. 2019 Jul 9;47(12):6195-6207. doi: 10.1093/nar/gkz374. Nucleic Acids Res. 2019. PMID: 31114898 Free PMC article.
-
Quantitative imaging of chromatin decompaction in living cells.Mol Biol Cell. 2018 Jul 15;29(13):1763-1777. doi: 10.1091/mbc.E17-11-0648. Epub 2018 May 17. Mol Biol Cell. 2018. PMID: 29771637 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
