

Diagnosis features of pediatric Gaucher disease patients in the era of enzymatic therapy, a national-base study from the Spanish Registry of Gaucher Disease
- PMID: 28468677
- PMCID: PMC5415726
- DOI: 10.1186/s13023-017-0627-z
Diagnosis features of pediatric Gaucher disease patients in the era of enzymatic therapy, a national-base study from the Spanish Registry of Gaucher Disease
Abstract
Background: The enzymatic replacement therapy (ERT) availability for Gaucher disease (GD) has changed the landscape of the disease, several countries have screening programs. These actions have promoted the early diagnosis and avoided many complications in pediatric patients. In Spain ERT has been available since 1993 and 386 patients have been included in the Spanish Registry of Gaucher Disease (SpRGD). The aim of this study is to analyze the impact of ERT on the characteristics at time of diagnosis and initial complications in pediatric Gaucher disease patients.
Aim: To analyze the impact of ERT on the characteristics at time of diagnosis and initial complications in pediatric Gaucher disease patients.
Methods: A review of data in SpRGD from patients' diagnosed before 18 years old was performed. The cohort was split according the year of diagnosis (≤1994, cohort A; ≥1995, cohort B).
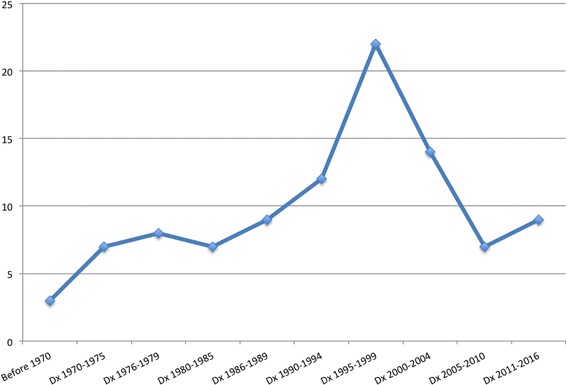
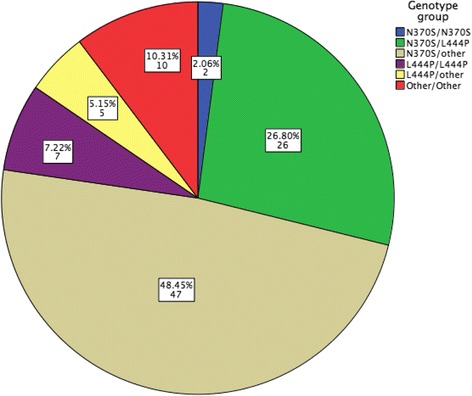
Results: A total of 98 pediatric patients were included, GD1: 80, GD3: 18; mean age: 7.2 (0.17-16.5) years, 58 (59.2%) males and 40 (40.8%) females. Forty-five were diagnosed ≤ 1994 and 53 ≥ 1995. Genotype: N370S/N370S: 2 (2.0%), N370S/L444P: 27 (27.5%), N370S/other: 47 (48%), L444P/L444P: 7 (7.1%), L444P/D409H: 2 (2.0%), L444P/other: 3 (6.2%), other/other: 10 (10.2%). The mean age at diagnosis was earlier in patients diagnosed after 1995 (p < 0.001) and different between the subtypes, GD1: 8.2 (0.2-16.5) years and GD3: 2.8 (0.17-10.2) years (p < 0.001). There were more severe patients in the group diagnosed before 1994 (p = 0.045) carrying L444P (2), D409H (2), G377S (1), G195W (1) or the recombinant mutation. The patients' diagnosed ≤1994 showed worse cytopenias, higher chance of bone vascular complications at diagnosis and previous spleen removal. The patients started ERT at a median time after diagnosis of 5.2 years [cohort A] and 1.6 years [cohort B] (p < 0.001).
Conclusions: The early diagnosis of Gaucher disease in the era of ERT availability has permitted to reduce the incidence of severe and irreversible initial complication in pediatric patients, and this has permitted better development of these patients. This is the largest pediatric cohort from a national registry.
Keywords: Children; Enzymatic replacement therapy; Gaucher Disease.
Figures
References
-
- Mehta A. Gaucher disease. Lijec Vjesn. 2007;129(Suppl 3):37. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
