

Whole-Genome Restriction Mapping by "Subhaploid"-Based RAD Sequencing: An Efficient and Flexible Approach for Physical Mapping and Genome Scaffolding
- PMID: 28468906
- PMCID: PMC5500127
- DOI: 10.1534/genetics.117.200303
Whole-Genome Restriction Mapping by "Subhaploid"-Based RAD Sequencing: An Efficient and Flexible Approach for Physical Mapping and Genome Scaffolding
Abstract
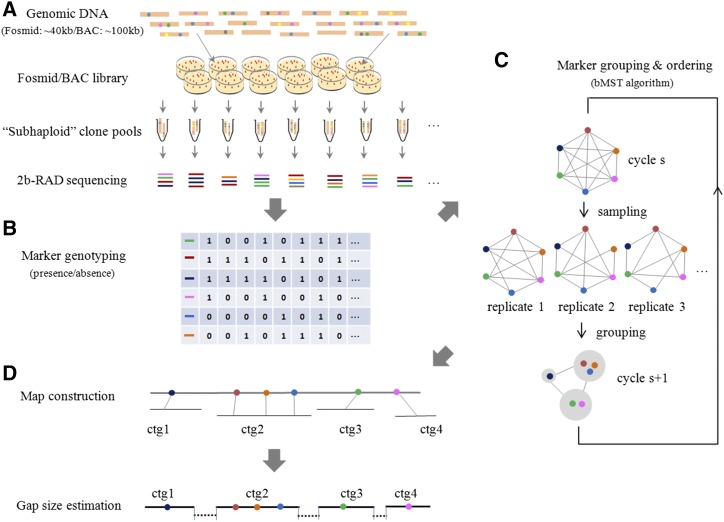
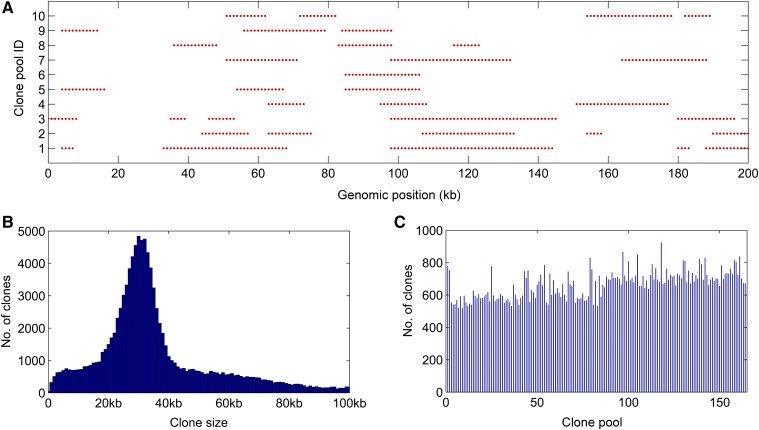
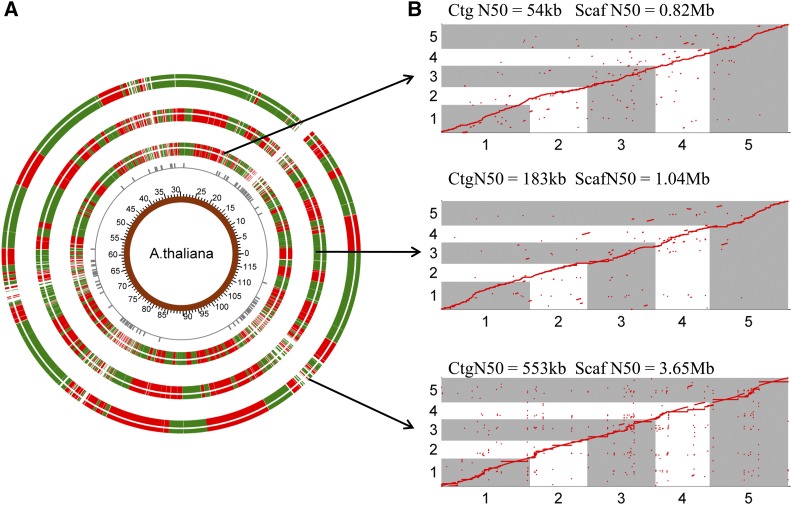
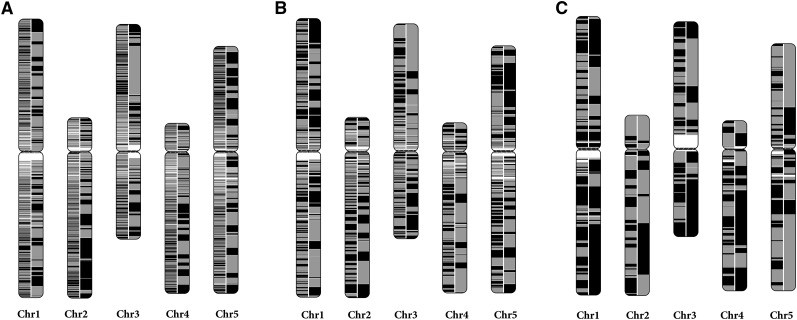
Assembly of complex genomes using short reads remains a major challenge, which usually yields highly fragmented assemblies. Generation of ultradense linkage maps is promising for anchoring such assemblies, but traditional linkage mapping methods are hindered by the infrequency and unevenness of meiotic recombination that limit attainable map resolution. Here we develop a sequencing-based "in vitro" linkage mapping approach (called RadMap), where chromosome breakage and segregation are realized by generating hundreds of "subhaploid" fosmid/bacterial-artificial-chromosome clone pools, and by restriction site-associated DNA sequencing of these clone pools to produce an ultradense whole-genome restriction map to facilitate genome scaffolding. A bootstrap-based minimum spanning tree algorithm is developed for grouping and ordering of genome-wide markers and is implemented in a user-friendly, integrated software package (AMMO). We perform extensive analyses to validate the power and accuracy of our approach in the model plant Arabidopsis thaliana and human. We also demonstrate the utility of RadMap for enhancing the contiguity of a variety of whole-genome shotgun assemblies generated using either short Illumina reads (300 bp) or long PacBio reads (6-14 kb), with up to 15-fold improvement of N50 (∼816 kb-3.7 Mb) and high scaffolding accuracy (98.1-98.5%). RadMap outperforms BioNano and Hi-C when input assembly is highly fragmented (contig N50 = 54 kb). RadMap can capture wide-range contiguity information and provide an efficient and flexible tool for high-resolution physical mapping and scaffolding of highly fragmented assemblies.
Keywords: RAD sequencing; genome scaffolding; in vitro linkage mapping; restriction map.
Copyright © 2017 Dou et al.
Figures
Similar articles
-
MaGuS: a tool for quality assessment and scaffolding of genome assemblies with Whole Genome Profiling™ Data.BMC Bioinformatics. 2016 Mar 3;17:115. doi: 10.1186/s12859-016-0969-x. BMC Bioinformatics. 2016. PMID: 26936254 Free PMC article.
-
Improving and correcting the contiguity of long-read genome assemblies of three plant species using optical mapping and chromosome conformation capture data.Genome Res. 2017 May;27(5):778-786. doi: 10.1101/gr.213652.116. Epub 2017 Feb 3. Genome Res. 2017. PMID: 28159771 Free PMC article.
-
BioNano genome mapping of individual chromosomes supports physical mapping and sequence assembly in complex plant genomes.Plant Biotechnol J. 2016 Jul;14(7):1523-31. doi: 10.1111/pbi.12513. Epub 2016 Jan 23. Plant Biotechnol J. 2016. PMID: 26801360 Free PMC article.
-
Using optical mapping data for the improvement of vertebrate genome assemblies.Gigascience. 2015 Mar 18;4:10. doi: 10.1186/s13742-015-0052-y. eCollection 2015. Gigascience. 2015. PMID: 25789164 Free PMC article. Review.
-
Genetic anchoring of whole-genome shotgun assemblies.Front Genet. 2014 Jul 7;5:208. doi: 10.3389/fgene.2014.00208. eCollection 2014. Front Genet. 2014. PMID: 25071835 Free PMC article. Review.
Cited by
-
Including diverse and admixed populations in genetic epidemiology research.Genet Epidemiol. 2022 Oct;46(7):347-371. doi: 10.1002/gepi.22492. Epub 2022 Jul 16. Genet Epidemiol. 2022. PMID: 35842778 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
