

Modelling amyotrophic lateral sclerosis: progress and possibilities
- PMID: 28468939
- PMCID: PMC5451175
- DOI: 10.1242/dmm.029058
Modelling amyotrophic lateral sclerosis: progress and possibilities
Abstract
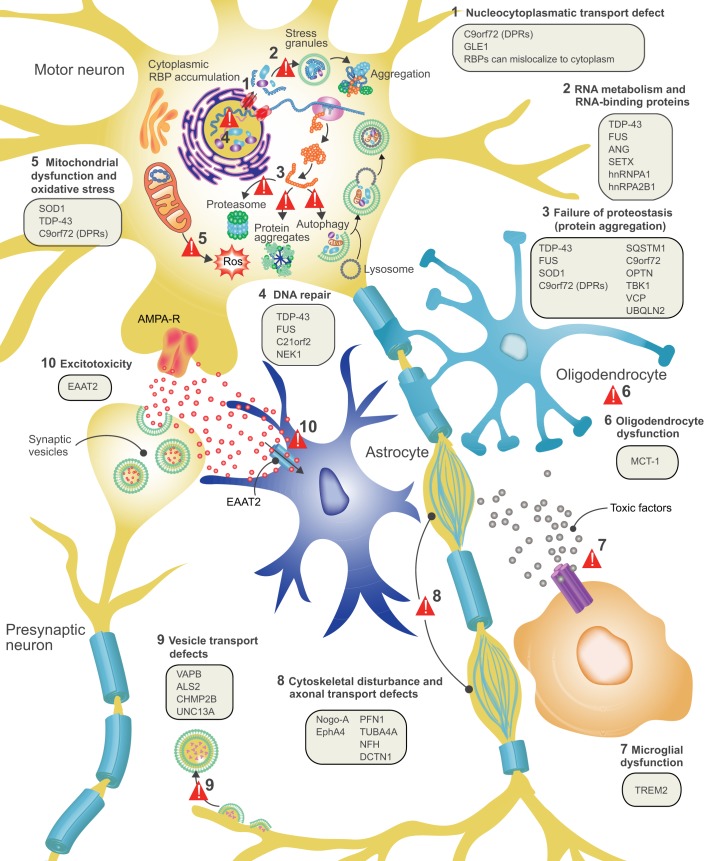
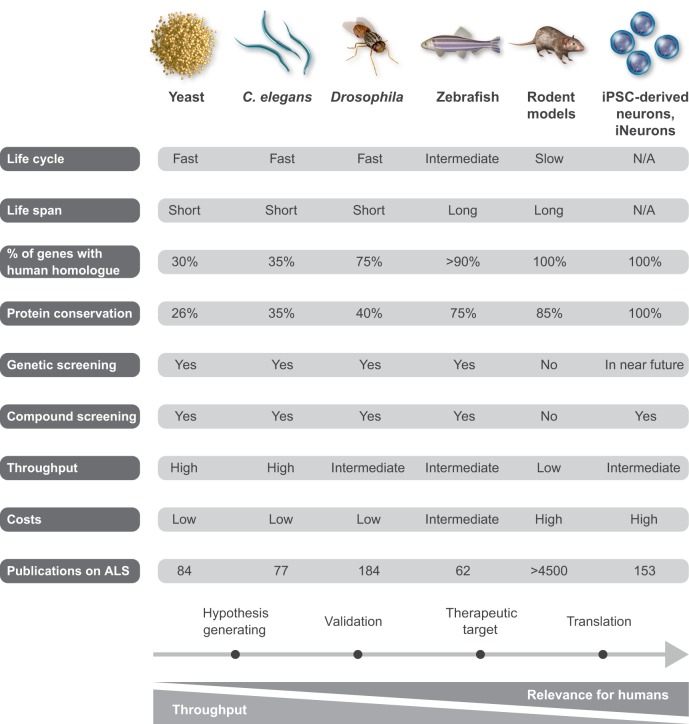
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder that primarily affects the motor system and presents with progressive muscle weakness. Most patients survive for only 2-5 years after disease onset, often due to failure of the respiratory muscles. ALS is a familial disease in ∼10% of patients, with the remaining 90% developing sporadic ALS. Over the past decade, major advances have been made in our understanding of the genetics and neuropathology of ALS. To date, around 20 genes are associated with ALS, with the most common causes of typical ALS associated with mutations in SOD1, TARDBP, FUS and C9orf72 Advances in our understanding of the genetic basis of ALS have led to the creation of different models of this disease. The molecular pathways that have emerged from these systems are more heterogeneous than previously anticipated, ranging from protein aggregation and defects in multiple key cellular processes in neurons, to dysfunction of surrounding non-neuronal cells. Here, we review the different model systems used to study ALS and discuss how they have contributed to our current knowledge of ALS disease mechanisms. A better understanding of emerging disease pathways, the detrimental effects of the various gene mutations and the causes underlying motor neuron denegation in sporadic ALS will accelerate progress in the development of novel treatments.
Keywords: C. elegans; Fruit fly; Motor neuron; Neurodegeneration; Zebrafish; iPSCs.
© 2017. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interestsThe authors declare no competing or financial interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
