

Transcriptional dissection of melanoma identifies a high-risk subtype underlying TP53 family genes and epigenome deregulation
- PMID: 28469092
- PMCID: PMC5414564
- DOI: 10.1172/jci.insight.92102
Transcriptional dissection of melanoma identifies a high-risk subtype underlying TP53 family genes and epigenome deregulation
Abstract
Background: Melanoma is a heterogeneous malignancy. We set out to identify the molecular underpinnings of high-risk melanomas, those that are likely to progress rapidly, metastasize, and result in poor outcomes.
Methods: We examined transcriptome changes from benign states to early-, intermediate-, and late-stage tumors using a set of 78 treatment-naive melanocytic tumors consisting of primary melanomas of the skin and benign melanocytic lesions. We utilized a next-generation sequencing platform that enabled a comprehensive analysis of protein-coding and -noncoding RNA transcripts.
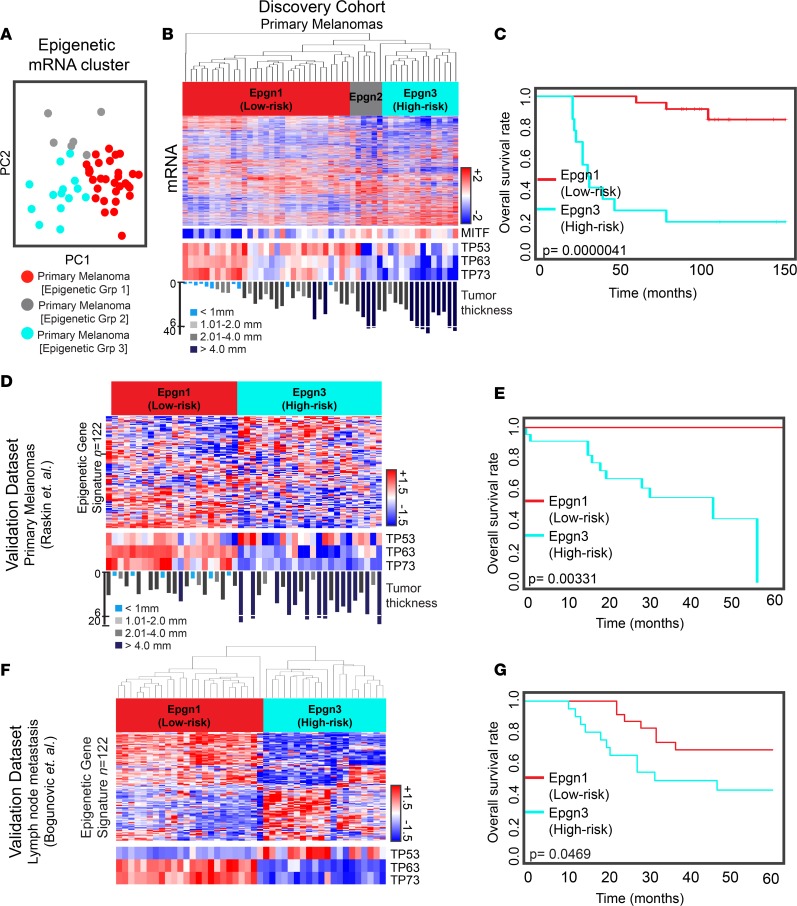
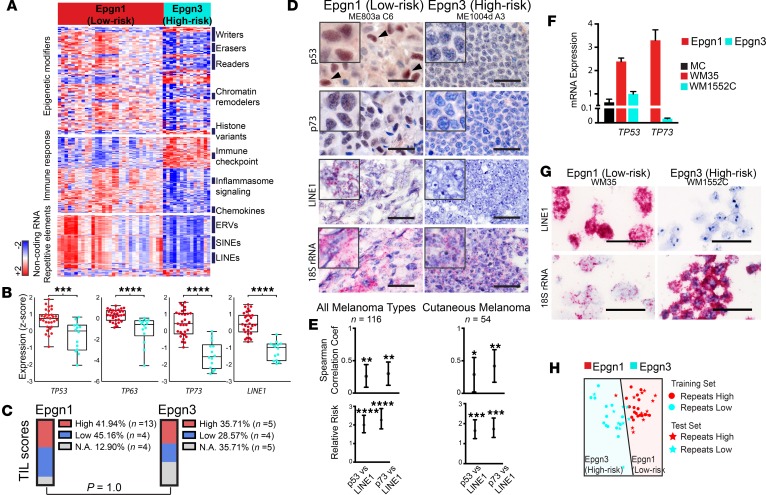
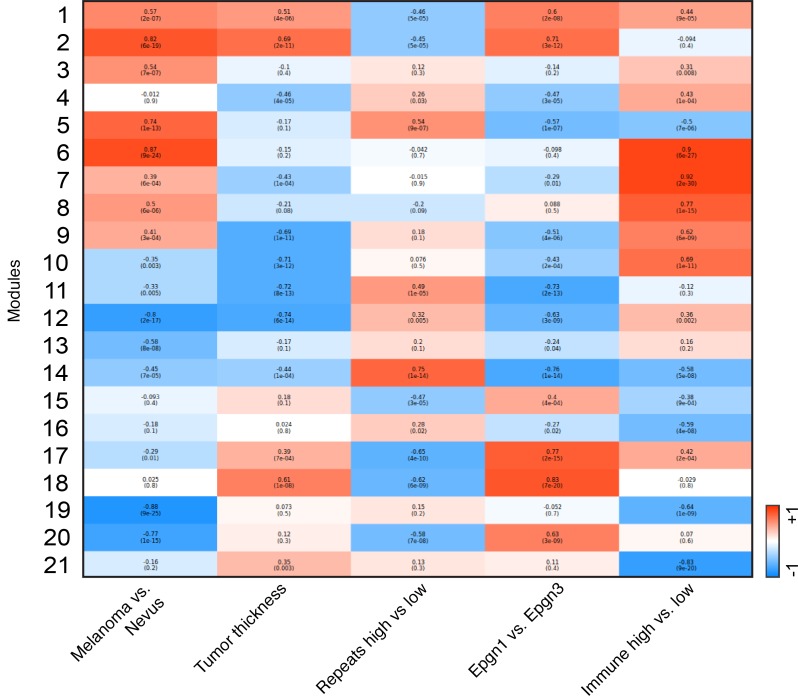
Results: Gene expression changes unequivocally discriminated between benign and malignant states, and a dual epigenetic and immune signature emerged defining this transition. To our knowledge, we discovered previously unrecognized melanoma subtypes. A high-risk primary melanoma subset was distinguished by a 122-epigenetic gene signature ("epigenetic" cluster) and TP53 family gene deregulation (TP53, TP63, and TP73). This subtype associated with poor overall survival and showed enrichment of cell cycle genes. Noncoding repetitive element transcripts (LINEs, SINEs, and ERVs) that can result in immunostimulatory signals recapitulating a state of "viral mimicry" were significantly repressed. The high-risk subtype and its poor predictive characteristics were validated in several independent cohorts. Additionally, primary melanomas distinguished by specific immune signatures ("immune" clusters) were identified.
Conclusion: The TP53 family of genes and genes regulating the epigenetic machinery demonstrate strong prognostic and biological relevance during progression of early disease. Gene expression profiling of protein-coding and -noncoding RNA transcripts may be a better predictor for disease course in melanoma. This study outlines the transcriptional interplay of the cancer cell's epigenome with the immune milieu with potential for future therapeutic targeting.
Funding: National Institutes of Health (CA154683, CA158557, CA177940, CA087497-13), Tisch Cancer Institute, Melanoma Research Foundation, the Dow Family Charitable Foundation, and the Icahn School of Medicine at Mount Sinai.
Keywords: Dermatology; Oncology.
Conflict of interest statement
Figures



References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
