

Amino Acid Interaction (INTAA) web server
- PMID: 28472475
- PMCID: PMC5570164
- DOI: 10.1093/nar/gkx352
Amino Acid Interaction (INTAA) web server
Abstract
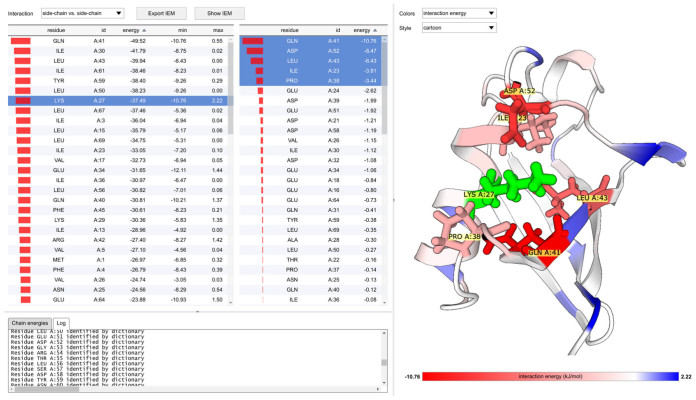
Large biomolecules-proteins and nucleic acids-are composed of building blocks which define their identity, properties and binding capabilities. In order to shed light on the energetic side of interactions of amino acids between themselves and with deoxyribonucleotides, we present the Amino Acid Interaction web server (http://bioinfo.uochb.cas.cz/INTAA/). INTAA offers the calculation of the residue Interaction Energy Matrix for any protein structure (deposited in Protein Data Bank or submitted by the user) and a comprehensive analysis of the interfaces in protein-DNA complexes. The Interaction Energy Matrix web application aims to identify key residues within protein structures which contribute significantly to the stability of the protein. The application provides an interactive user interface enhanced by 3D structure viewer for efficient visualization of pairwise and net interaction energies of individual amino acids, side chains and backbones. The protein-DNA interaction analysis part of the web server allows the user to view the relative abundance of various configurations of amino acid-deoxyribonucleotide pairs found at the protein-DNA interface and the interaction energies corresponding to these configurations calculated using a molecular mechanical force field. The effects of the sugar-phosphate moiety and of the dielectric properties of the solvent on the interaction energies can be studied for the various configurations.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

References
-
- Bendova-Biedermannova L., Hobza P., Vondrasek J.. Identifying stabilizing key residues in proteins using interresidue interaction energy matrix. Proteins. 2008; 72:402–413. - PubMed
-
- Vondrasek J., Bendova L., Klusak V., Hobza P.. Unexpectedly strong energy stabilization inside the hydrophobic core of small protein rubredoxin mediated by aromatic residues: correlated ab initio quantum chemical calculations. J. Am. Chem. Soc. 2005; 127:2615–2619. - PubMed
-
- Kaminski G.A., Friesner R.A., Tirado-Rives J., Jorgensen W.L.. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001; 105:6474–6487.
-
- Duan Y., Wu C., Chowdhury S., Lee M.C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T. et al. . A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003; 24:1999–2012. - PubMed
-
- Berka K., Laskowski R.A., Hobza P., Vondrasek J.. Energy matrix of structurally important side-chain/side-chain interactions in proteins. J. Chem. Theory Comput. 2010; 6:2191–2203. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
