

Janus Kinase 3, a Novel Regulator for Smooth Muscle Proliferation and Vascular Remodeling
- PMID: 28473442
- PMCID: PMC5503700
- DOI: 10.1161/ATVBAHA.116.308895
Janus Kinase 3, a Novel Regulator for Smooth Muscle Proliferation and Vascular Remodeling
Abstract
Objective: Vascular remodeling because of smooth muscle cell (SMC) proliferation is a common process occurring in several vascular diseases, such as atherosclerosis, aortic aneurysm, post-transplant vasculopathy, restenosis after angioplasty, etc. The molecular mechanism underlying SMC proliferation, however, is not completely understood. The objective of this study is to determine the role and mechanism of Janus kinase 3 (JAK3) in vascular remodeling and SMC proliferation.
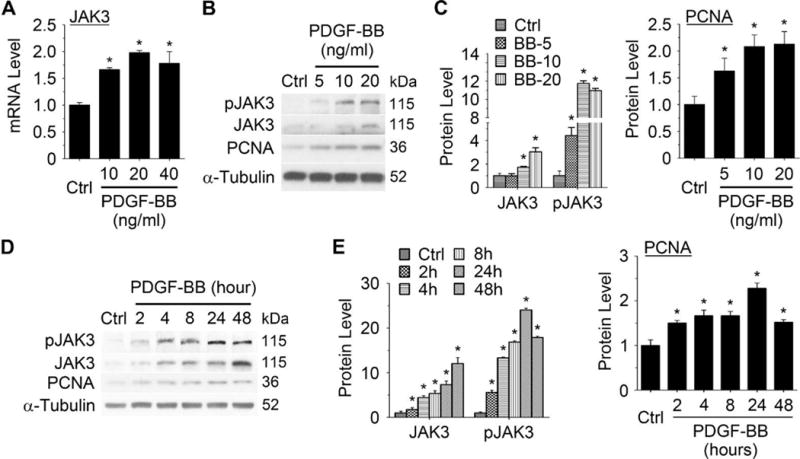
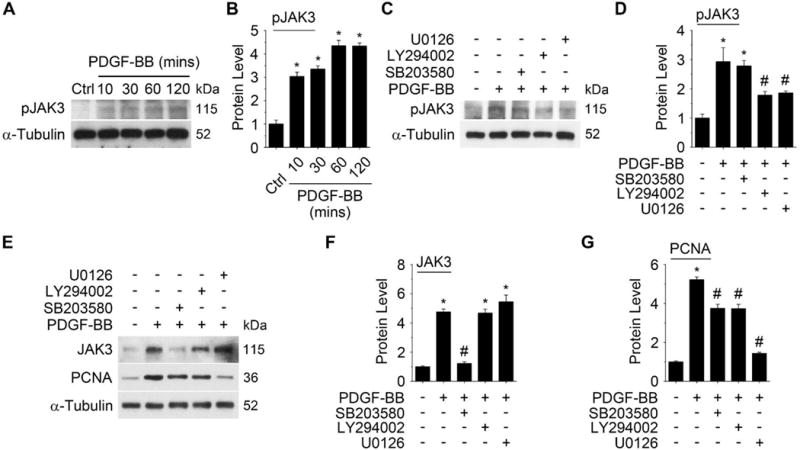
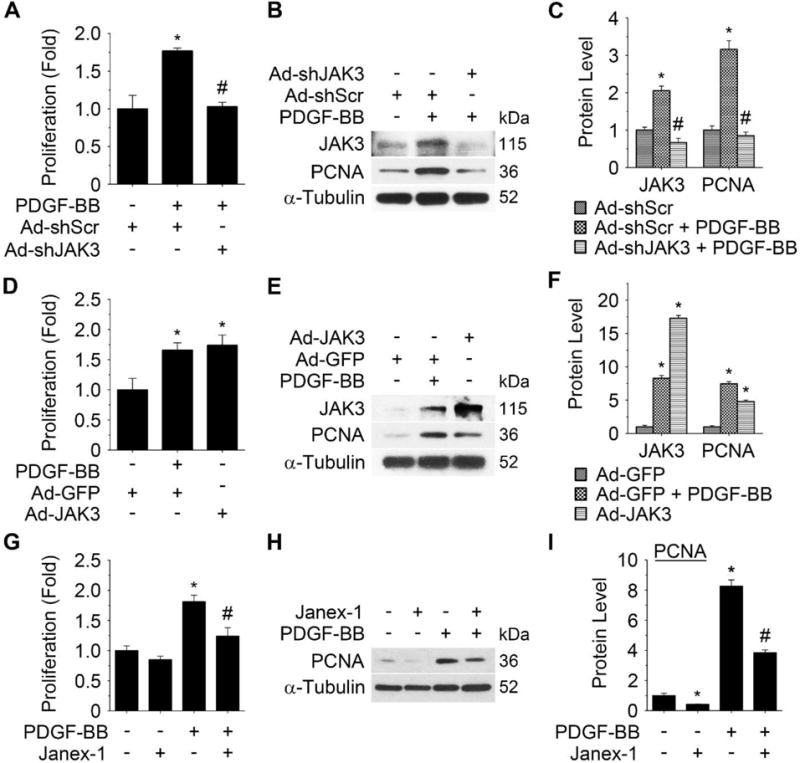
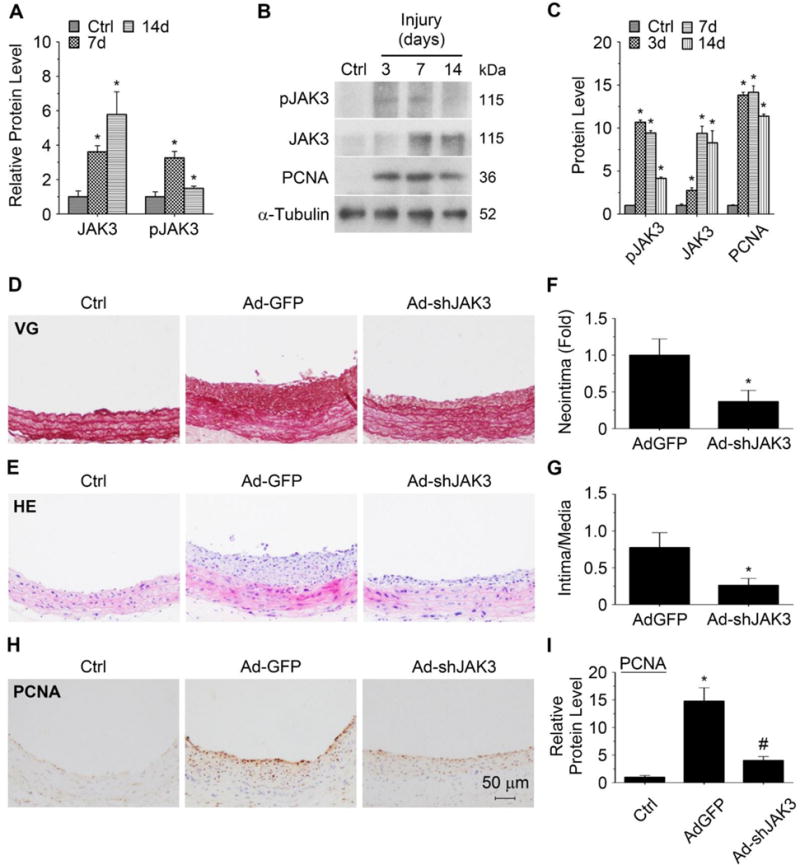
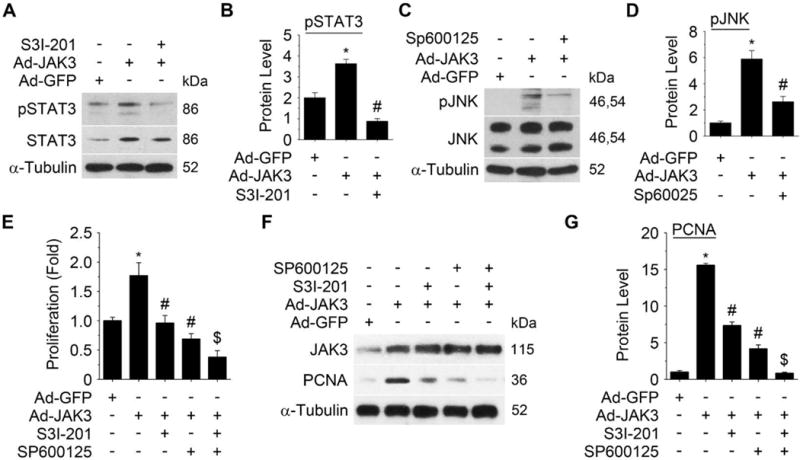
Approach and results: Platelet-derived growth factor-BB, an SMC mitogen, induces JAK3 expression and phosphorylation while stimulating SMC proliferation. Janex-1, a specific inhibitor of JAK3, or knockdown of JAK3 by short hairpin RNA, inhibits the SMC proliferation. Conversely, ectopic expression of JAK3 promotes SMC proliferation. Mechanistically, JAK3 promotes the phosphorylation of signal transducer and activator of transcription 3 and c-Jun N-terminal kinase in SMC, 2 signaling pathways known to be critical for SMC proliferation and vascular remodeling. Blockade of these 2 signaling pathways by their inhibitors impeded the JAK3-mediated SMC proliferation. In vivo, knockdown of JAK3 attenuates injury-induced neointima formation with attenuated neointimal SMC proliferation. Knockdown of JAK3 also induces neointimal SMC apoptosis in rat carotid artery balloon injury model.
Conclusions: Our results demonstrate that JAK3 mediates SMC proliferation and survival during injury-induced vascular remodeling, which provides a potential therapeutic target for preventing neointimal hyperplasia in proliferative vascular diseases.
Keywords: Janus kinase 3; apoptosis; interleukin; smooth muscle proliferation; vascular remodeling.
© 2017 American Heart Association, Inc.
Figures

Similar articles
-
Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling.Circ Res. 2015 May 8;116(10):e71-80. doi: 10.1161/CIRCRESAHA.116.305863. Epub 2015 Mar 18. Circ Res. 2015. PMID: 25788409 Free PMC article.
-
Olfactomedin 2 Regulates Smooth Muscle Phenotypic Modulation and Vascular Remodeling Through Mediating Runt-Related Transcription Factor 2 Binding to Serum Response Factor.Arterioscler Thromb Vasc Biol. 2017 Mar;37(3):446-454. doi: 10.1161/ATVBAHA.116.308606. Epub 2017 Jan 5. Arterioscler Thromb Vasc Biol. 2017. PMID: 28062493 Free PMC article.
-
Soluble epoxide hydrolase is involved in the development of atherosclerosis and arterial neointima formation by regulating smooth muscle cell migration.Am J Physiol Heart Circ Physiol. 2015 Dec 1;309(11):H1894-903. doi: 10.1152/ajpheart.00289.2015. Epub 2015 Oct 9. Am J Physiol Heart Circ Physiol. 2015. PMID: 26453326
-
Inflammation and vascular injury: basic discovery to drug development.Circ J. 2012;76(8):1811-8. doi: 10.1253/circj.cj-12-0801. Epub 2012 Jul 30. Circ J. 2012. PMID: 22785436 Free PMC article. Review.
-
Direct role for smooth muscle cell mineralocorticoid receptors in vascular remodeling: novel mechanisms and clinical implications.Curr Hypertens Rep. 2014 May;16(5):427. doi: 10.1007/s11906-014-0427-y. Curr Hypertens Rep. 2014. PMID: 24633842 Free PMC article. Review.
Cited by
-
JANEX-1 improves acute pulmonary embolism through VEGF and FAK in pulmonary artery smooth muscle cells.Exp Biol Med (Maywood). 2020 Sep;245(15):1395-1403. doi: 10.1177/1535370220942474. Epub 2020 Jul 15. Exp Biol Med (Maywood). 2020. PMID: 32664806 Free PMC article.
-
BMPRII+ neural precursor cells isolated and characterized from organotypic neurospheres: an in vitro model of human fetal spinal cord development.Neural Regen Res. 2024 Feb;19(2):447-457. doi: 10.4103/1673-5374.373669. Neural Regen Res. 2024. PMID: 37488910 Free PMC article.
-
Interleukin-21 receptor gene polymorphism is associated with hepatitis B virus-related hepatocellular carcinoma in Chinese patients.J Clin Lab Anal. 2019 Jun;33(5):e22860. doi: 10.1002/jcla.22860. Epub 2019 Feb 13. J Clin Lab Anal. 2019. PMID: 30758075 Free PMC article.
-
Contribution of p62/SQSTM1 to PDGF-BB-induced myofibroblast-like phenotypic transition in vascular smooth muscle cells lacking Smpd1 gene.Cell Death Dis. 2018 Nov 19;9(12):1145. doi: 10.1038/s41419-018-1197-2. Cell Death Dis. 2018. PMID: 30451833 Free PMC article.
-
17β-Estradiol Inhibits Proliferation and Oxidative Stress in Vascular Smooth Muscle Cells by Upregulating BHLHE40 Expression.Front Cardiovasc Med. 2021 Nov 30;8:768662. doi: 10.3389/fcvm.2021.768662. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 34917665 Free PMC article.
References
-
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. - PubMed
-
- Verbsky JW, Bach EA, Fang YF, Yang L, Randolph DA, Fields LE. Expression of Janus kinase 3 in human endothelial and other non-lymphoid and non-myeloid cells. J Biol Chem. 1996;271:13976–80. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
