

Impact of cytosine methylation on DNA binding specificities of human transcription factors
- PMID: 28473536
- PMCID: PMC8009048
- DOI: 10.1126/science.aaj2239
Impact of cytosine methylation on DNA binding specificities of human transcription factors
Abstract
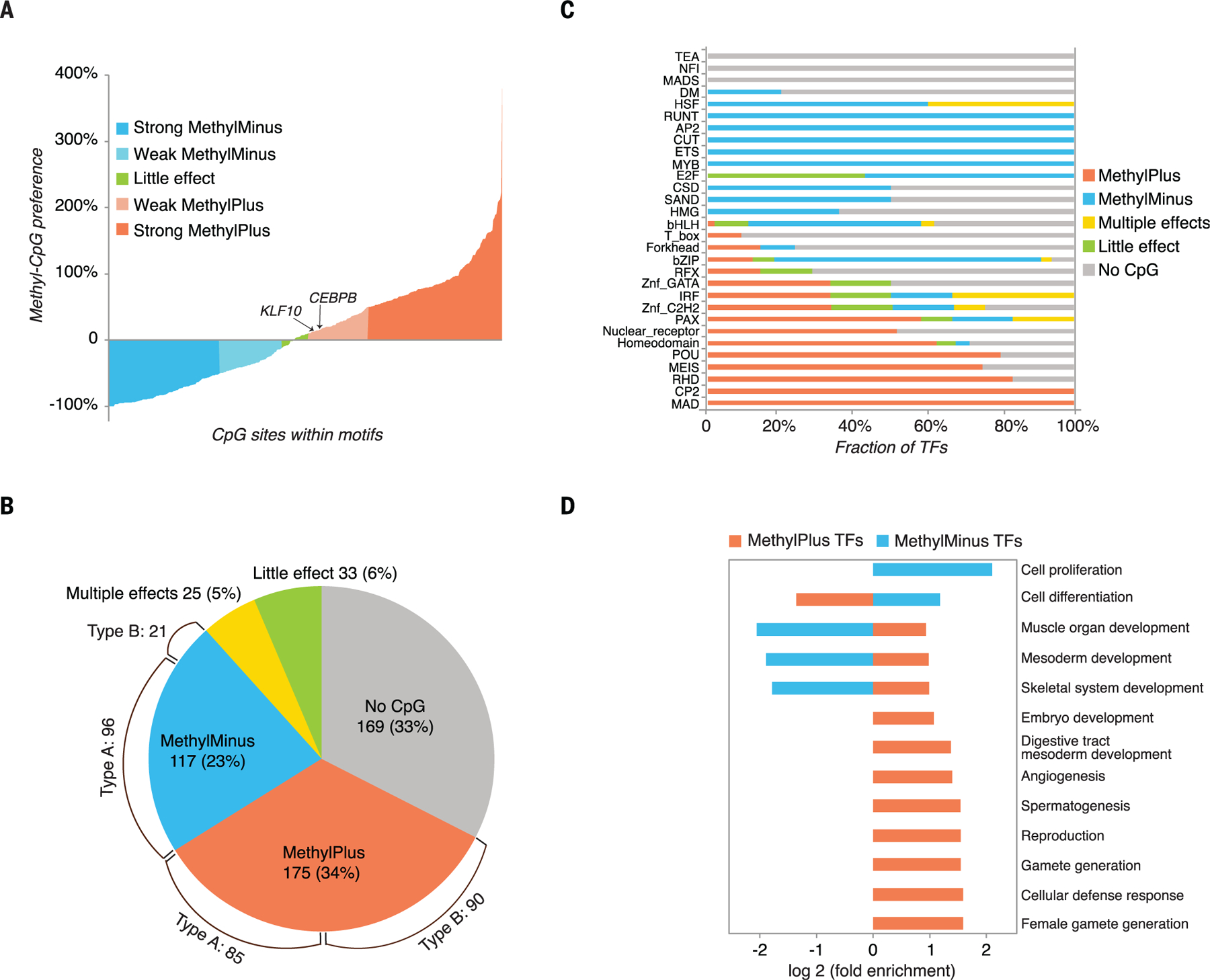
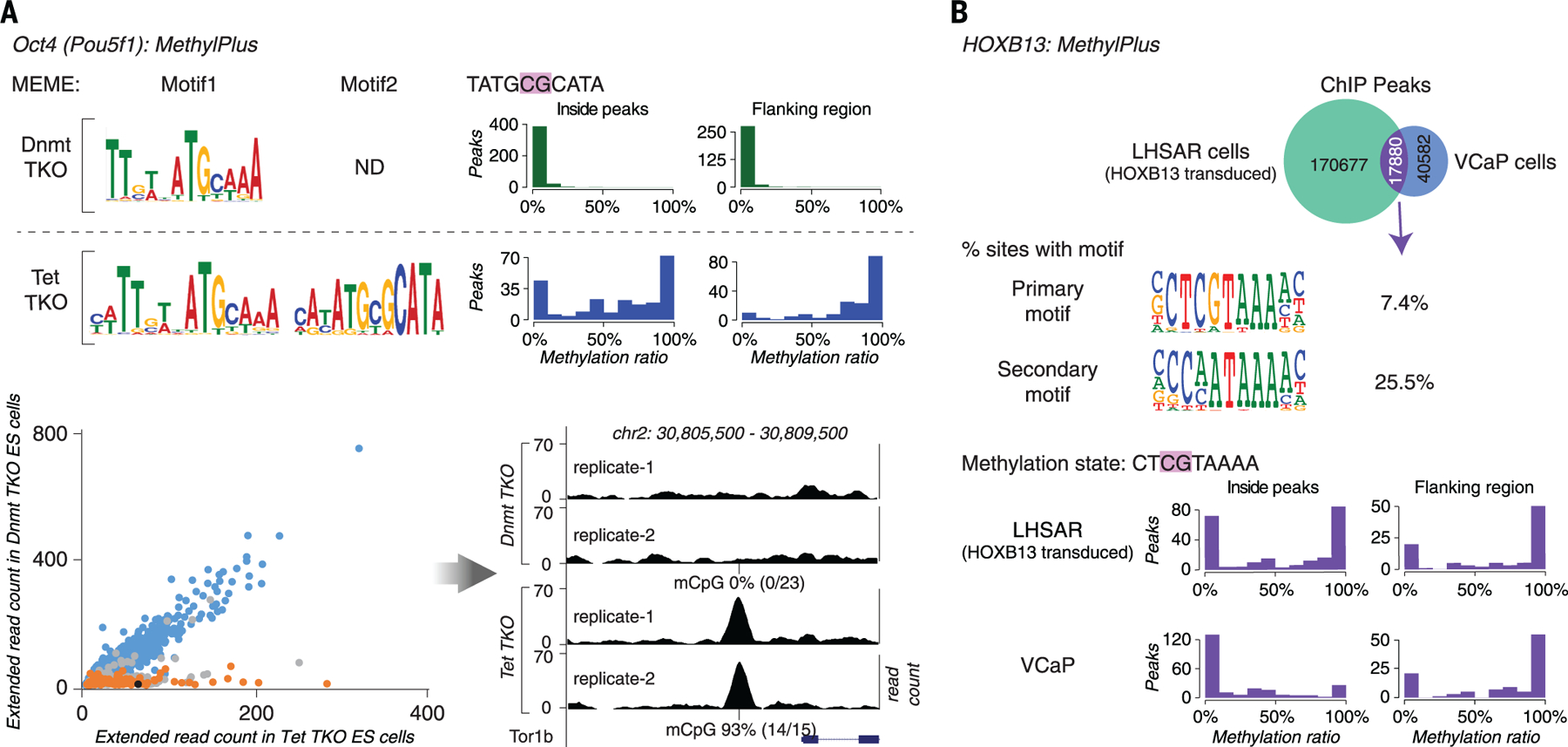
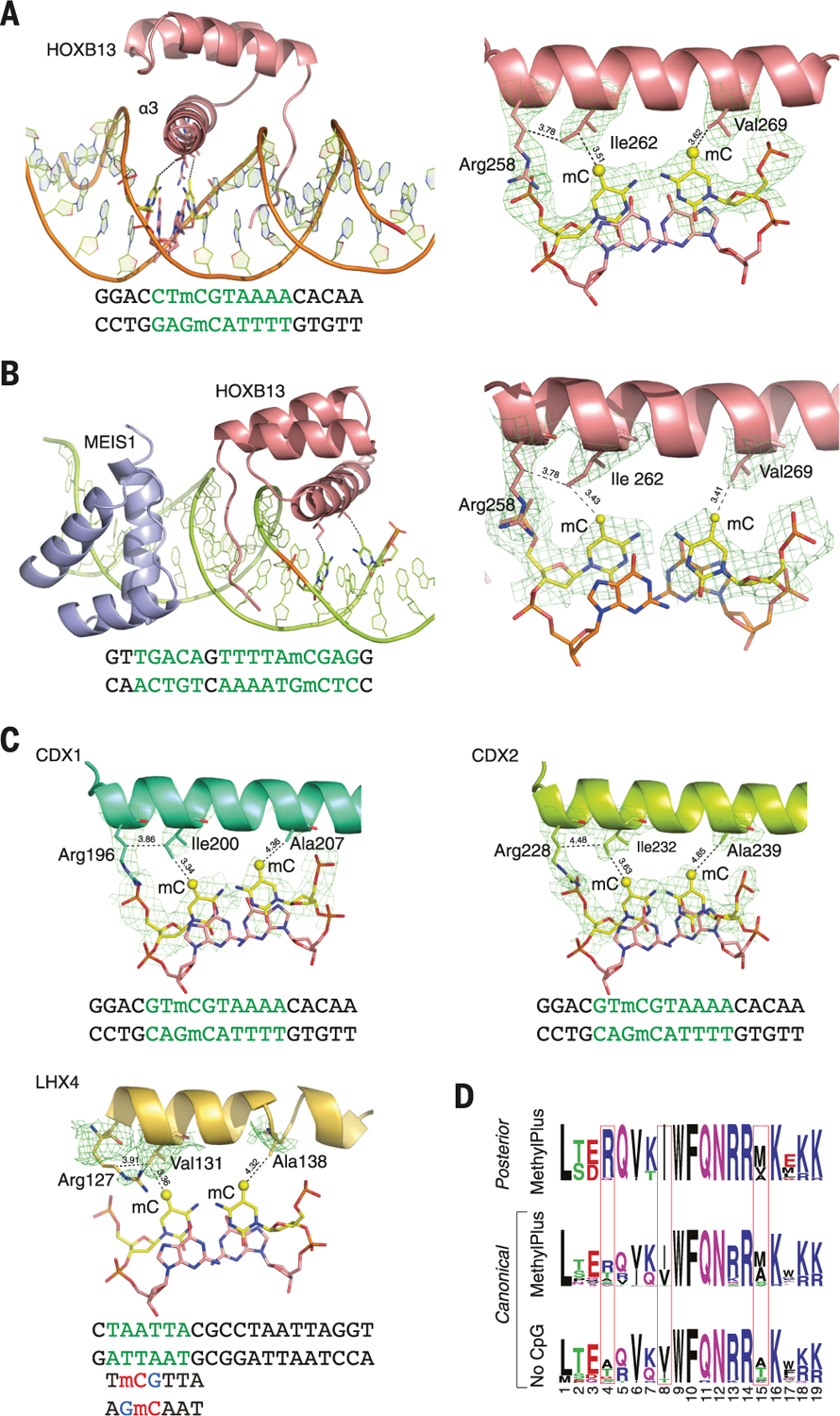
The majority of CpG dinucleotides in the human genome are methylated at cytosine bases. However, active gene regulatory elements are generally hypomethylated relative to their flanking regions, and the binding of some transcription factors (TFs) is diminished by methylation of their target sequences. By analysis of 542 human TFs with methylation-sensitive SELEX (systematic evolution of ligands by exponential enrichment), we found that there are also many TFs that prefer CpG-methylated sequences. Most of these are in the extended homeodomain family. Structural analysis showed that homeodomain specificity for methylcytosine depends on direct hydrophobic interactions with the methylcytosine 5-methyl group. This study provides a systematic examination of the effect of an epigenetic DNA modification on human TF binding specificity and reveals that many developmentally important proteins display preference for mCpG-containing sequences.
Copyright © 2017, American Association for the Advancement of Science.
Figures
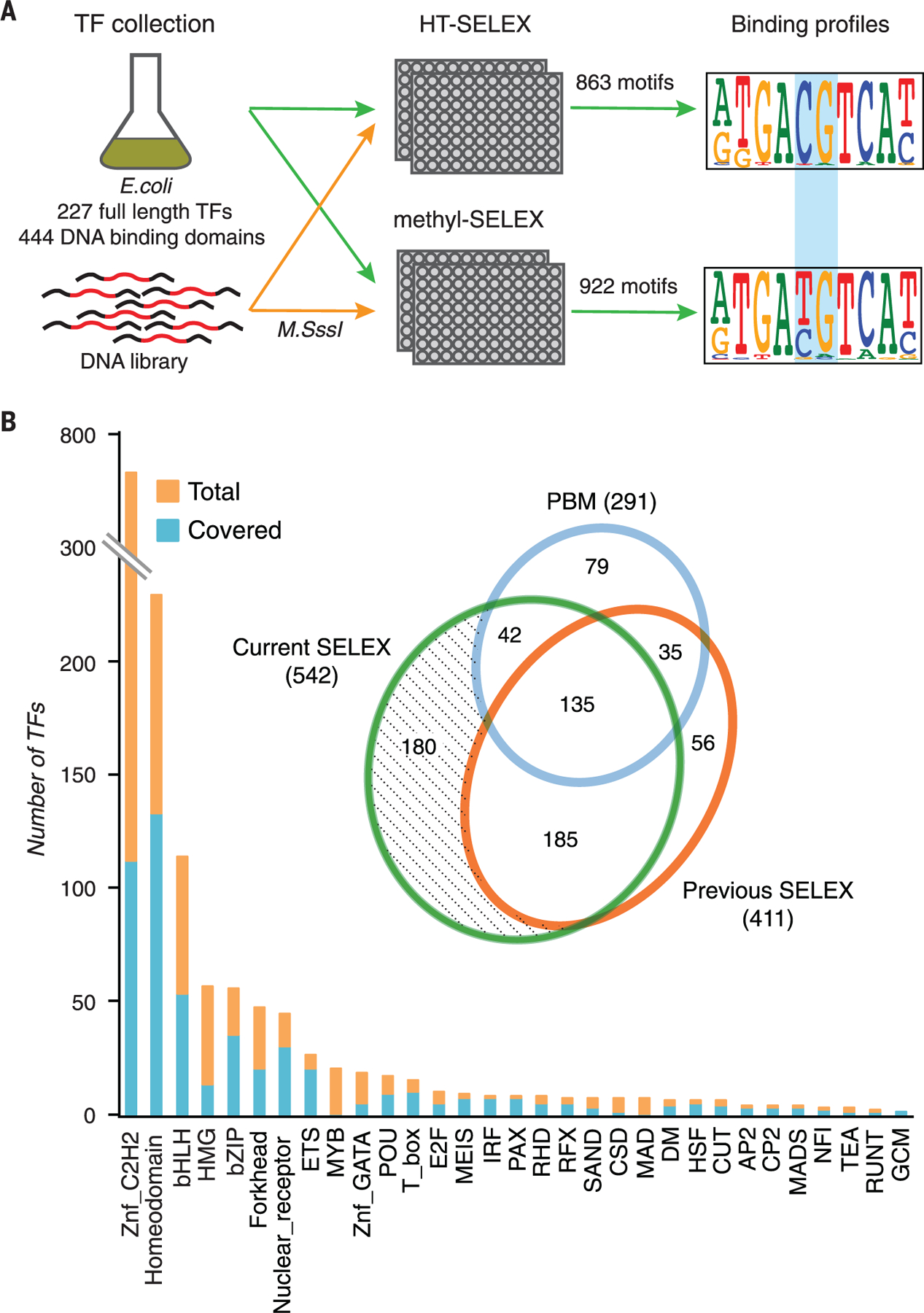
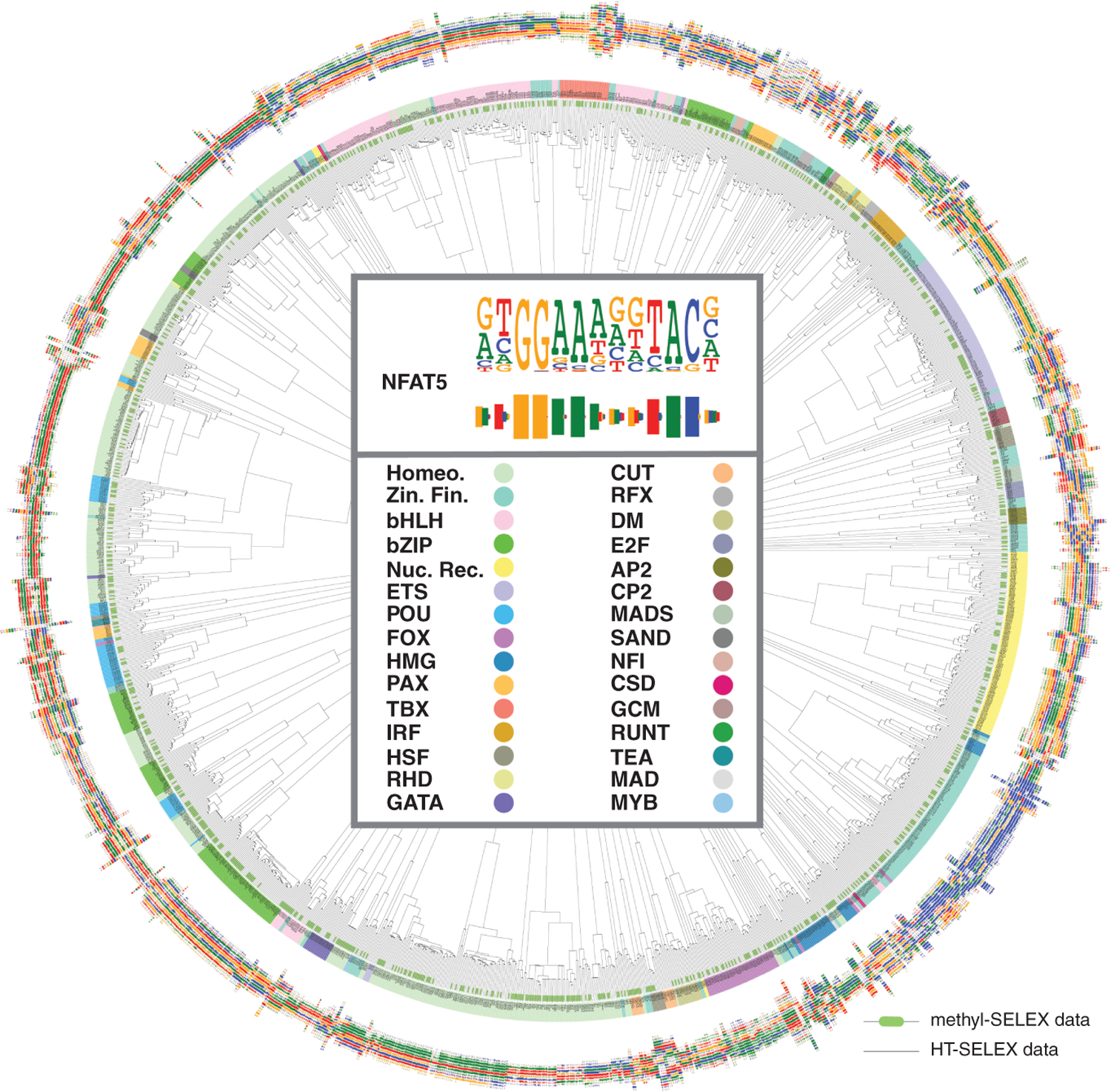
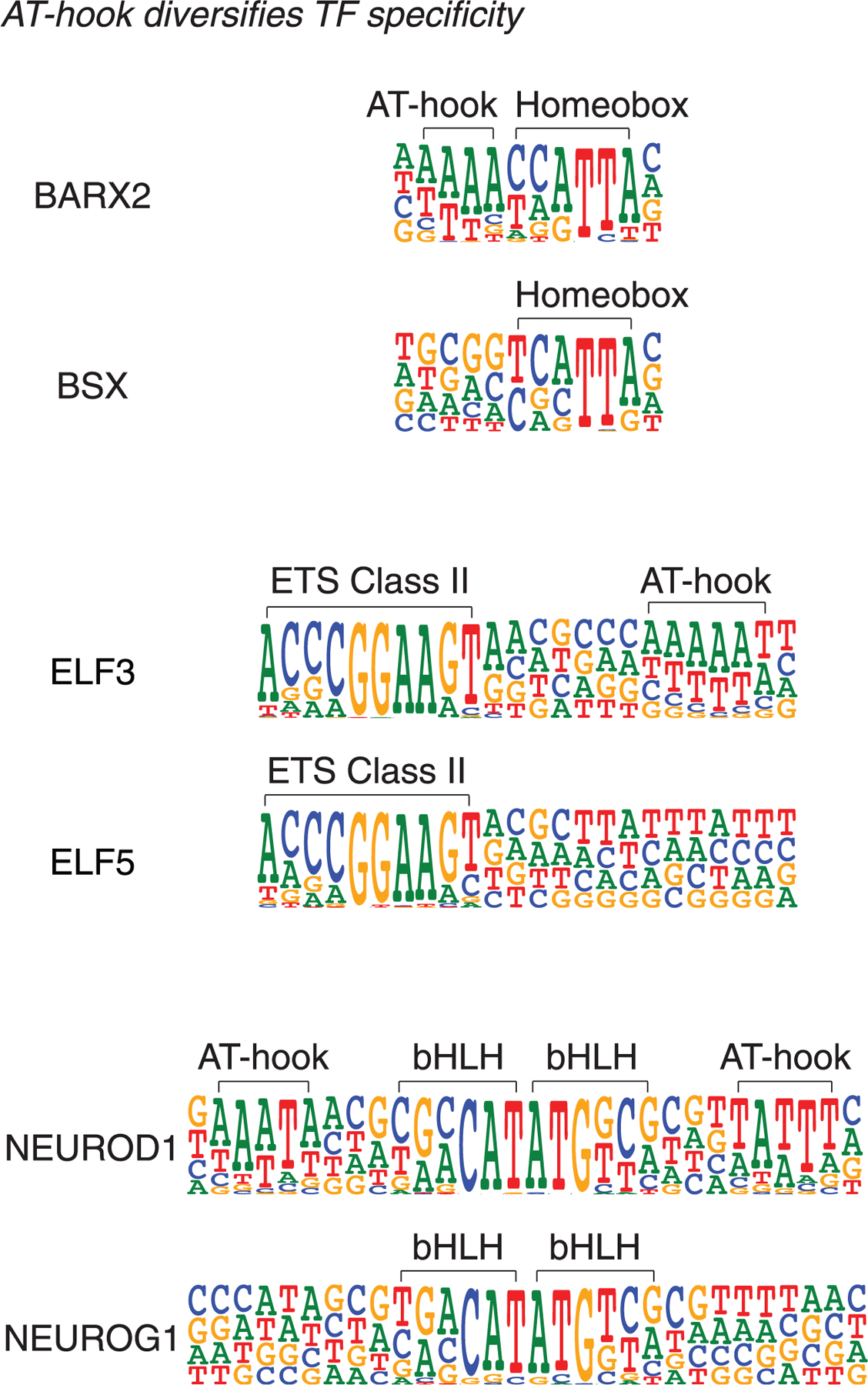
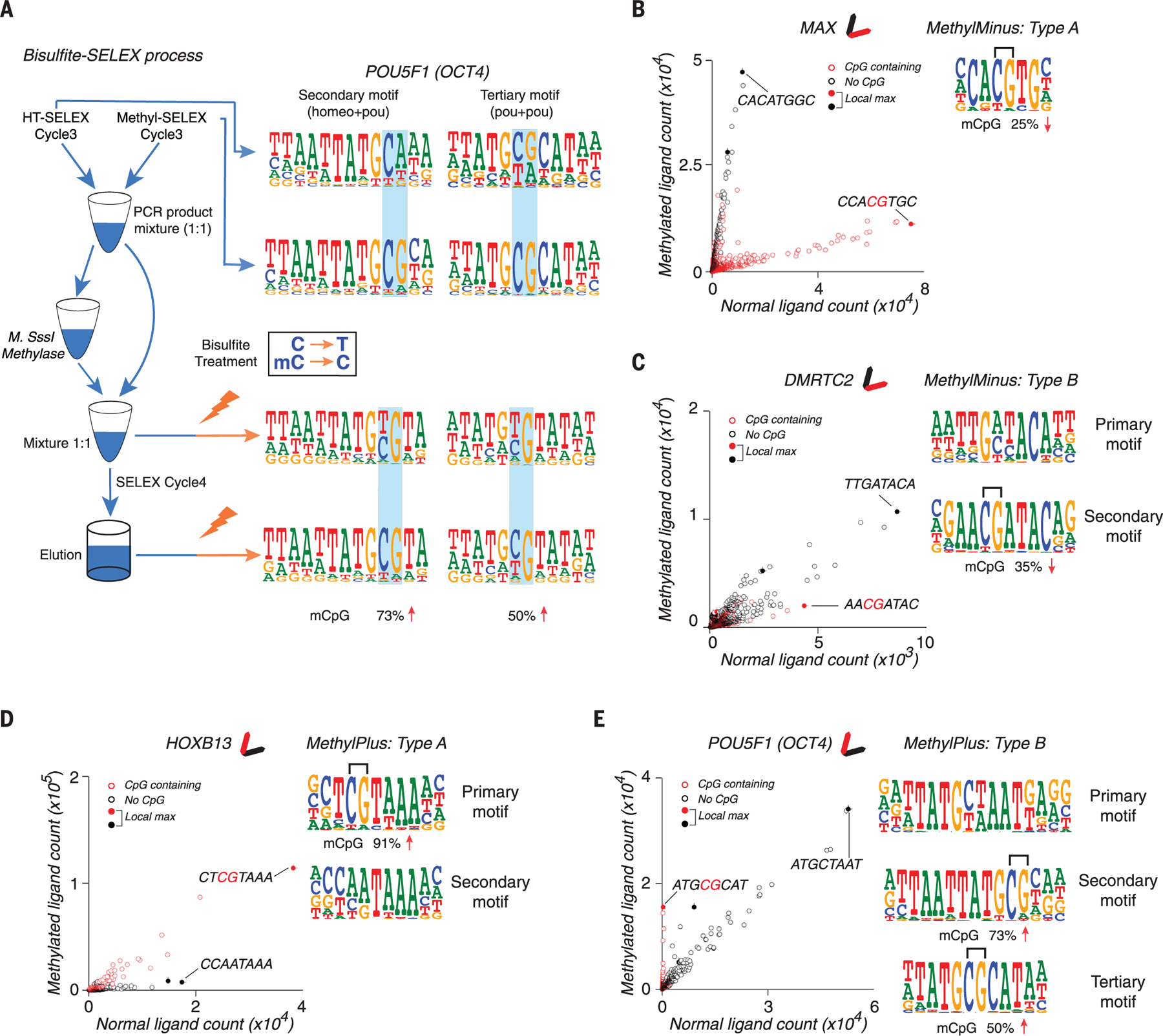
Comment in
-
Transcription factors read epigenetics.Science. 2017 May 5;356(6337):489-490. doi: 10.1126/science.aan2927. Science. 2017. PMID: 28473550 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
