

Quantification of differential gene expression by multiplexed targeted resequencing of cDNA
- PMID: 28474677
- PMCID: PMC5424154
- DOI: 10.1038/ncomms15190
Quantification of differential gene expression by multiplexed targeted resequencing of cDNA
Abstract
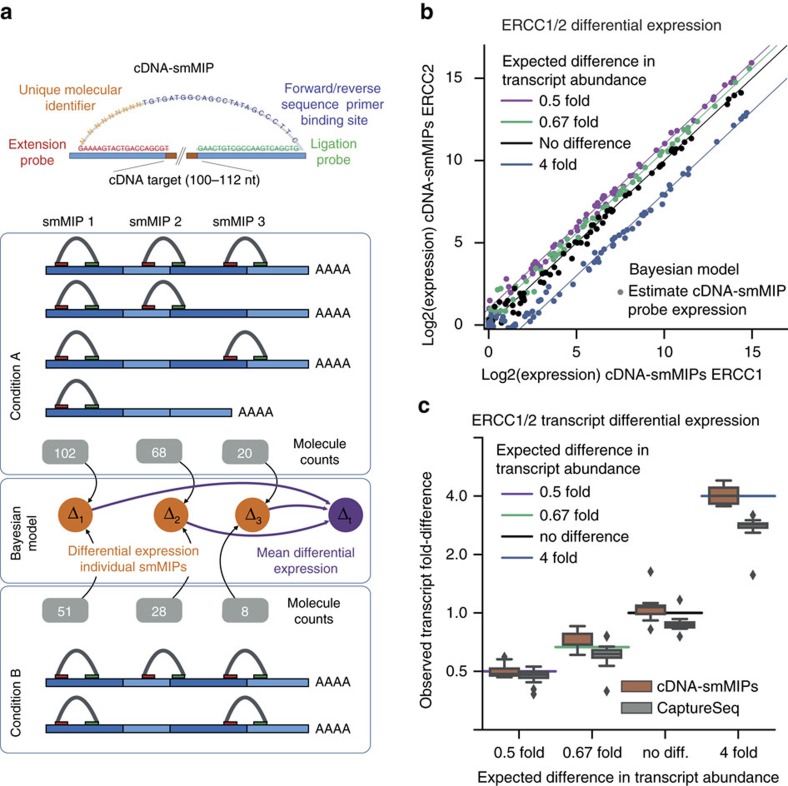
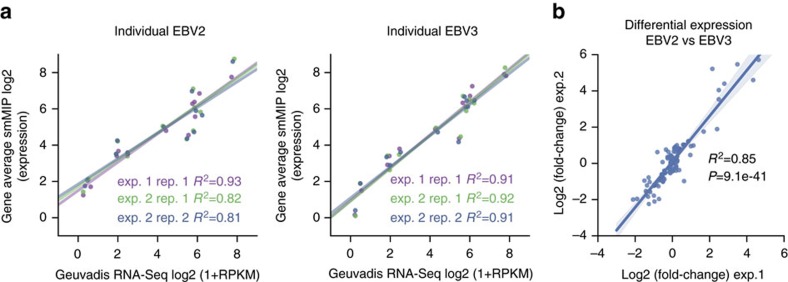
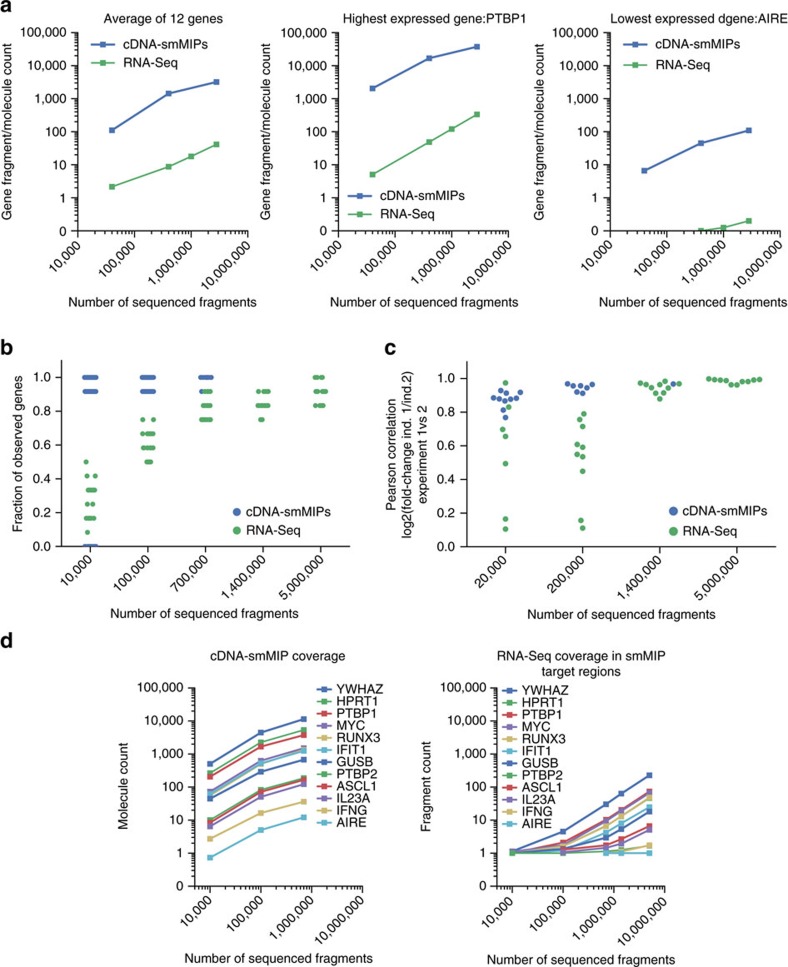
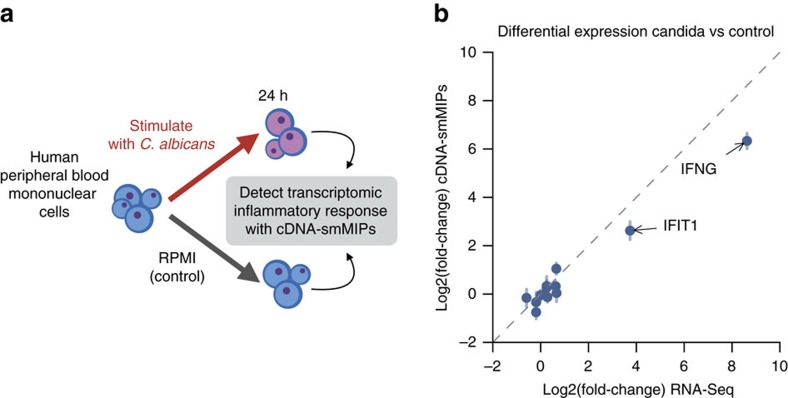
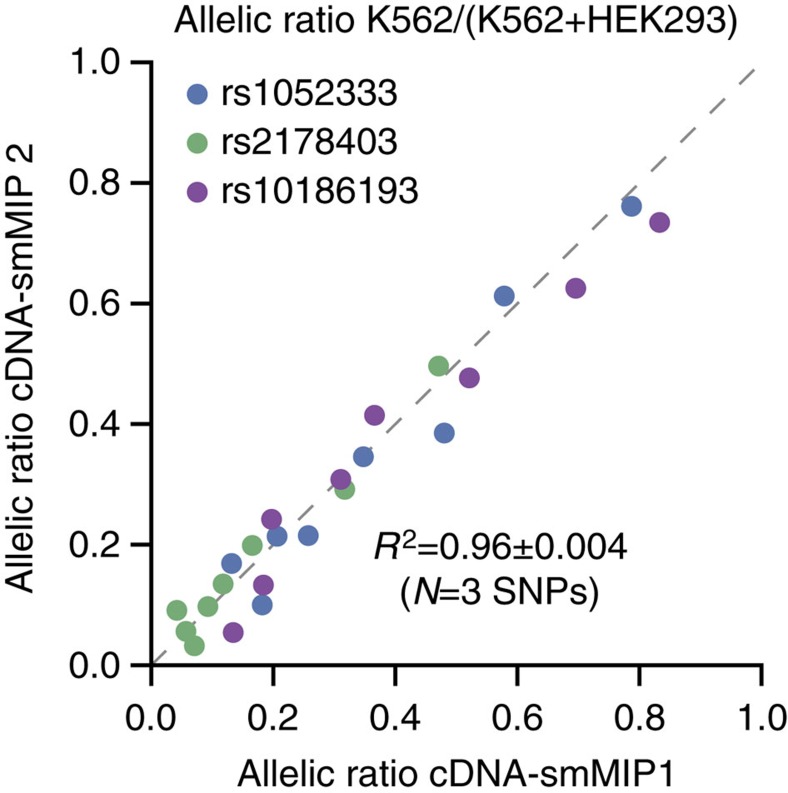
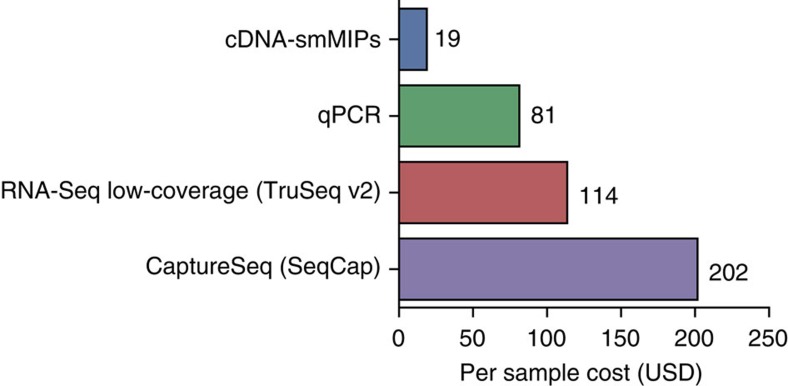
Whole-transcriptome or RNA sequencing (RNA-Seq) is a powerful and versatile tool for functional analysis of different types of RNA molecules, but sample reagent and sequencing cost can be prohibitive for hypothesis-driven studies where the aim is to quantify differential expression of a limited number of genes. Here we present an approach for quantification of differential mRNA expression by targeted resequencing of complementary DNA using single-molecule molecular inversion probes (cDNA-smMIPs) that enable highly multiplexed resequencing of cDNA target regions of ∼100 nucleotides and counting of individual molecules. We show that accurate estimates of differential expression can be obtained from molecule counts for hundreds of smMIPs per reaction and that smMIPs are also suitable for quantification of relative gene expression and allele-specific expression. Compared with low-coverage RNA-Seq and a hybridization-based targeted RNA-Seq method, cDNA-smMIPs are a cost-effective high-throughput tool for hypothesis-driven expression analysis in large numbers of genes (10 to 500) and samples (hundreds to thousands).
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Clark M. B. et al.. Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nat. Methods 12, 339–342 (2015). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
