

Peripheral complement interactions with amyloid β peptide: Erythrocyte clearance mechanisms
- PMID: 28475854
- PMCID: PMC5880643
- DOI: 10.1016/j.jalz.2017.03.010
Peripheral complement interactions with amyloid β peptide: Erythrocyte clearance mechanisms
Abstract
Introduction: Although amyloid β peptide (Aβ) is cleared from the brain to cerebrospinal fluid and the peripheral circulation, mechanisms for its removal from blood remain unresolved. Primates have uniquely evolved a highly effective peripheral clearance mechanism for pathogens, immune adherence, in which erythrocyte complement receptor 1 (CR1) plays a major role.
Methods: Multidisciplinary methods were used to demonstrate immune adherence capture of Aβ by erythrocytes and its deficiency in Alzheimer's disease (AD).
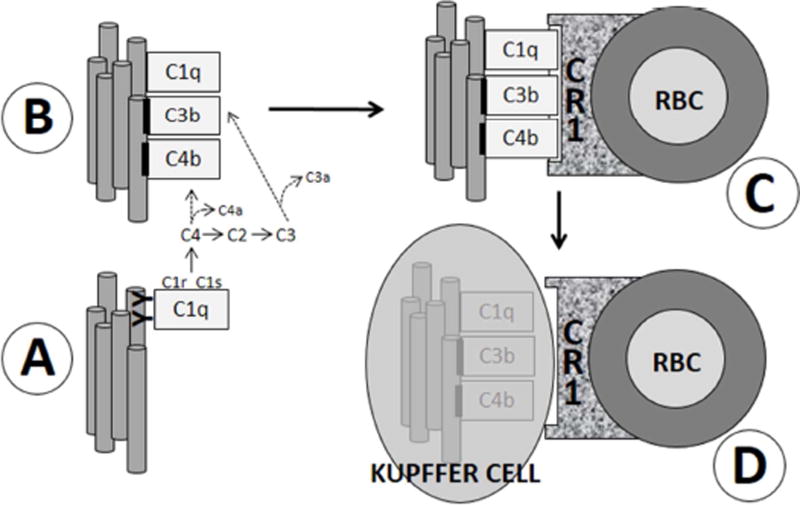
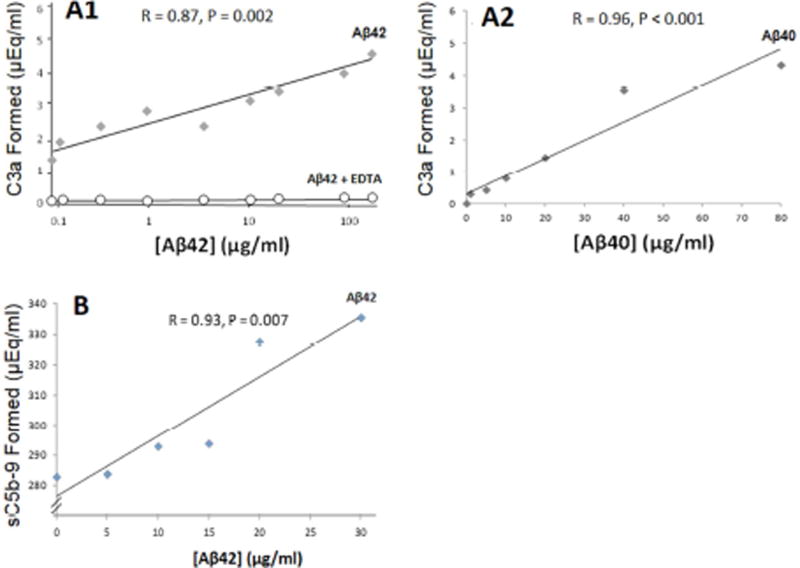
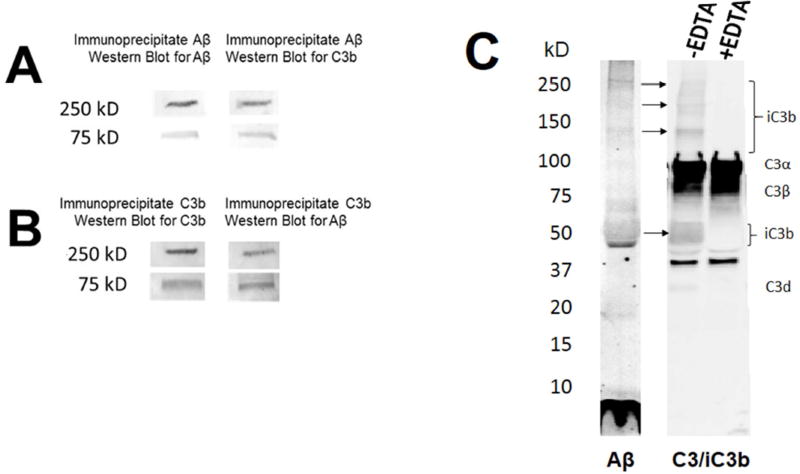
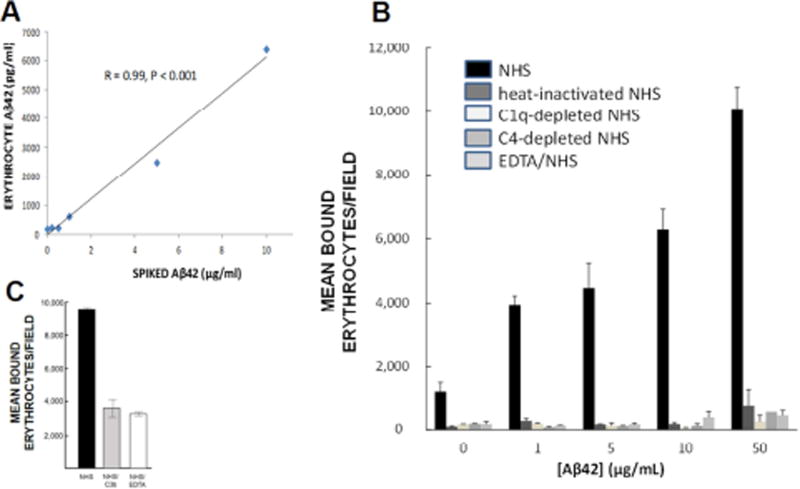
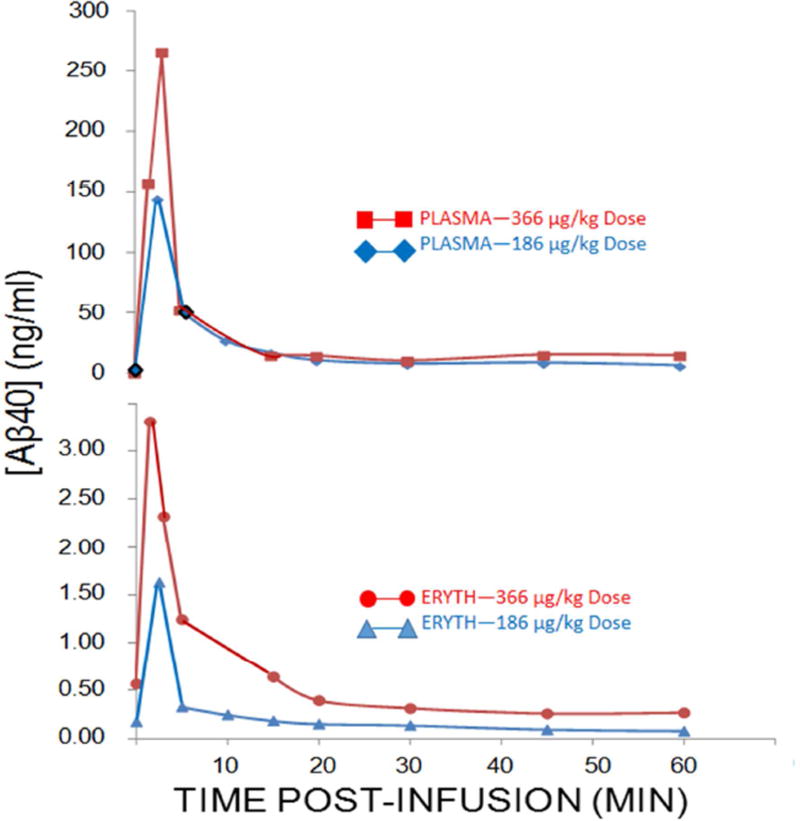
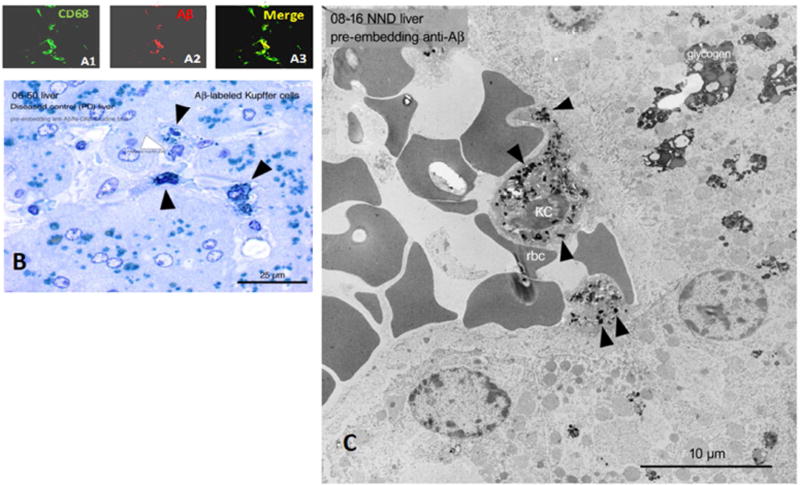
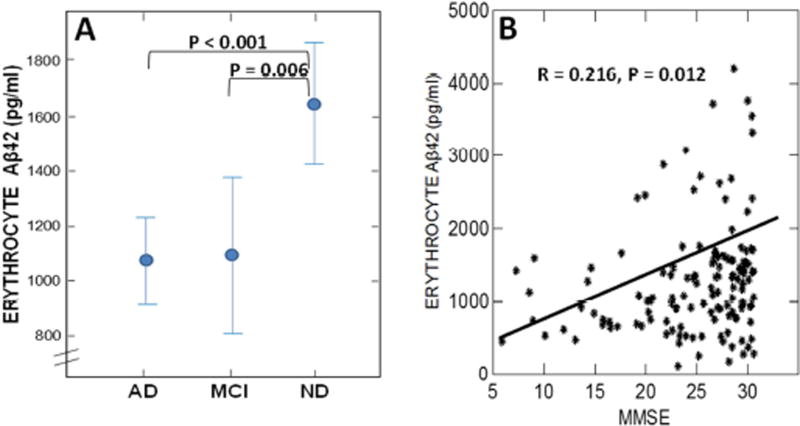
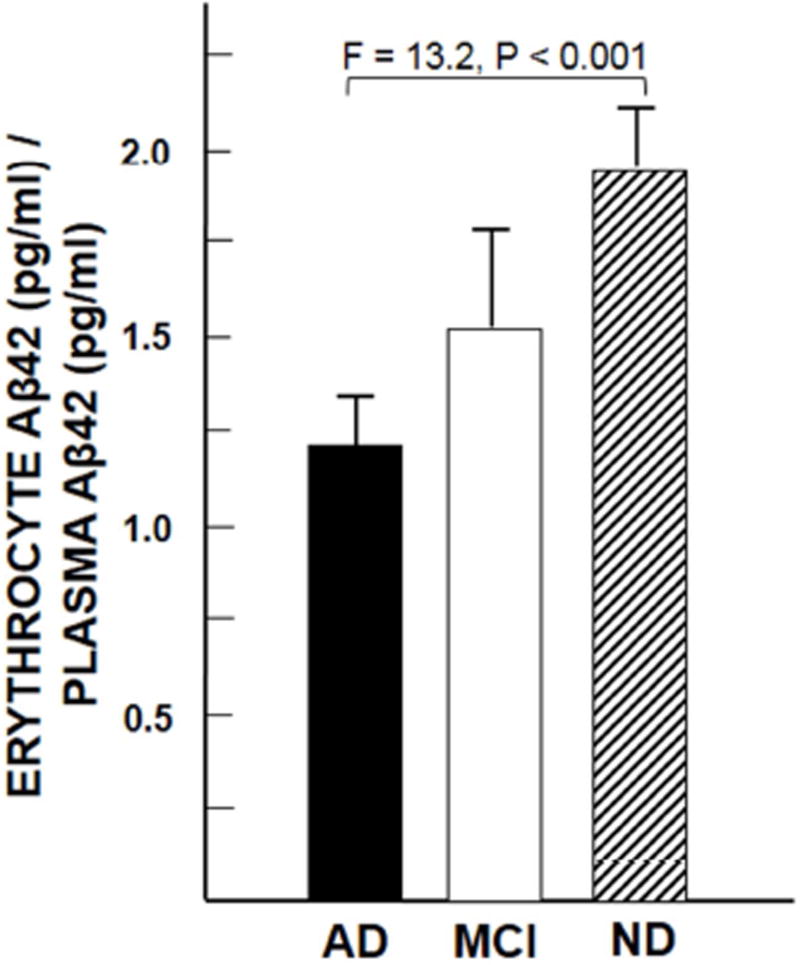
Results: Aβ was shown to be subject to immune adherence at every step in the pathway. Aβ dose-dependently activated serum complement. Complement-opsonized Aβ was captured by erythrocytes via CR1. Erythrocytes, Aβ, and hepatic Kupffer cells were colocalized in the human liver. Significant deficits in erythrocyte Aβ levels were found in AD and mild cognitive impairment patients.
Discussion: CR1 polymorphisms elevate AD risk, and >80% of human CR1 is vested in erythrocytes to subserve immune adherence. The present results suggest that this pathway is pathophysiologically relevant in AD.
Keywords: Alzheimer's disease; Amyloid β peptide; Blood; Complement; Complement receptor 1; Erythrocyte; Human; Immune adherence.
Copyright © 2017 the Alzheimer's Association. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
Similar articles
-
Peripheral complement interactions with amyloid β peptide in Alzheimer's disease: 2. Relationship to amyloid β immunotherapy.Alzheimers Dement. 2018 Feb;14(2):243-252. doi: 10.1016/j.jalz.2017.04.015. Epub 2017 Jul 26. Alzheimers Dement. 2018. PMID: 28755839 Free PMC article.
-
Peripheral complement interactions with amyloid β peptide in Alzheimer's disease: Polymorphisms, structure, and function of complement receptor 1.Alzheimers Dement. 2018 Nov;14(11):1438-1449. doi: 10.1016/j.jalz.2018.04.003. Epub 2018 May 21. Alzheimers Dement. 2018. PMID: 29792870 Free PMC article.
-
Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes.Neurobiol Aging. 2006 Dec;27(12):1733-9. doi: 10.1016/j.neurobiolaging.2005.09.043. Epub 2005 Nov 14. Neurobiol Aging. 2006. PMID: 16290270
-
Alzheimer's disease.Subcell Biochem. 2012;65:329-52. doi: 10.1007/978-94-007-5416-4_14. Subcell Biochem. 2012. PMID: 23225010 Review.
-
CR1 and CR1-like: the primate immune adherence receptors.Immunol Rev. 2001 Apr;180:100-11. doi: 10.1034/j.1600-065x.2001.1800109.x. Immunol Rev. 2001. PMID: 11414352 Review.
Cited by
-
Complement-Mediated Events in Alzheimer's Disease: Mechanisms and Potential Therapeutic Targets.J Immunol. 2020 Jan 15;204(2):306-315. doi: 10.4049/jimmunol.1901068. J Immunol. 2020. PMID: 31907273 Free PMC article. Review.
-
Subsequent correlated changes in complement component 3 and amyloid beta oligomers in the blood of patients with Alzheimer's disease.Alzheimers Dement. 2024 Apr;20(4):2731-2741. doi: 10.1002/alz.13734. Epub 2024 Feb 27. Alzheimers Dement. 2024. PMID: 38411315 Free PMC article.
-
Serum levels of proteins involved in amyloid-β clearance are related to cognitive decline and neuroimaging changes in mild cognitive impairment.Alzheimers Dement (Amst). 2019 Jan 12;11:85-97. doi: 10.1016/j.dadm.2018.11.003. eCollection 2019 Dec. Alzheimers Dement (Amst). 2019. PMID: 30671532 Free PMC article.
-
Extracellular protein components of amyloid plaques and their roles in Alzheimer's disease pathology.Mol Neurodegener. 2021 Aug 28;16(1):59. doi: 10.1186/s13024-021-00465-0. Mol Neurodegener. 2021. PMID: 34454574 Free PMC article. Review.
-
The good, the bad, and the opportunities of the complement system in neurodegenerative disease.J Neuroinflammation. 2020 Nov 25;17(1):354. doi: 10.1186/s12974-020-02024-8. J Neuroinflammation. 2020. PMID: 33239010 Free PMC article. Review.
References
-
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–13. - PubMed
-
- Do TM, Bedussi B, Chasseigneaux S, Dodacki A, Yapo C, Chacun H, et al. Oatp1a4 and an L-thyroxine-sensitive transporter mediate the mouse blood-brain barrier transport of amyloid-beta peptide. J Alzheimers Dis. 2013;36:555–61. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
