

Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability
- PMID: 28475857
- PMCID: PMC5420355
- DOI: 10.1016/j.ajhg.2017.03.010
Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability
Abstract
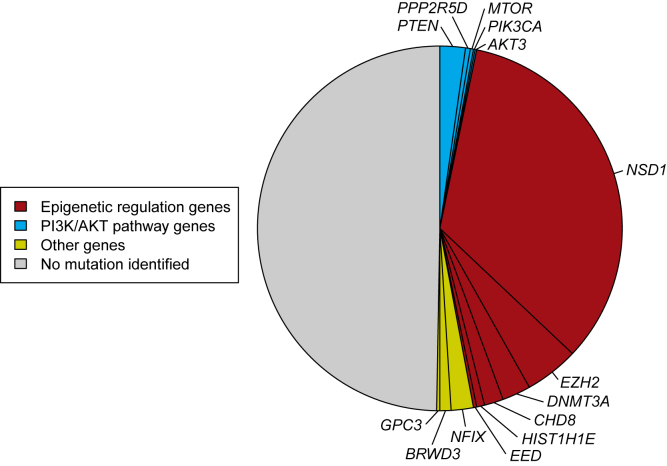
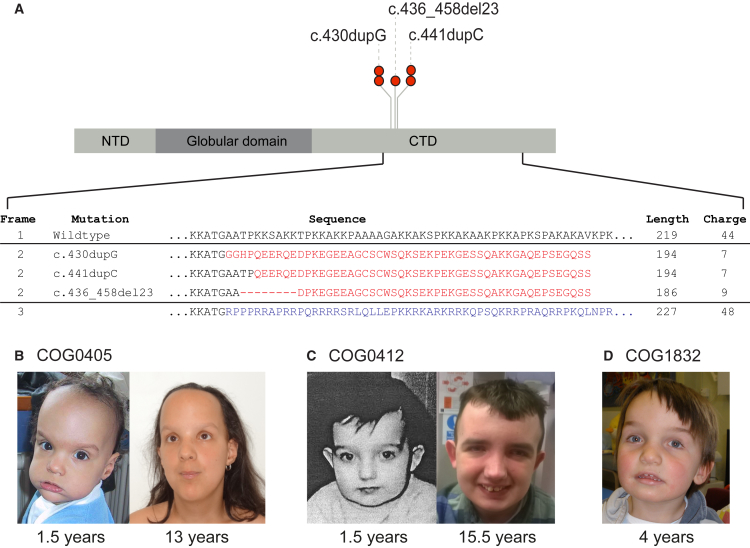
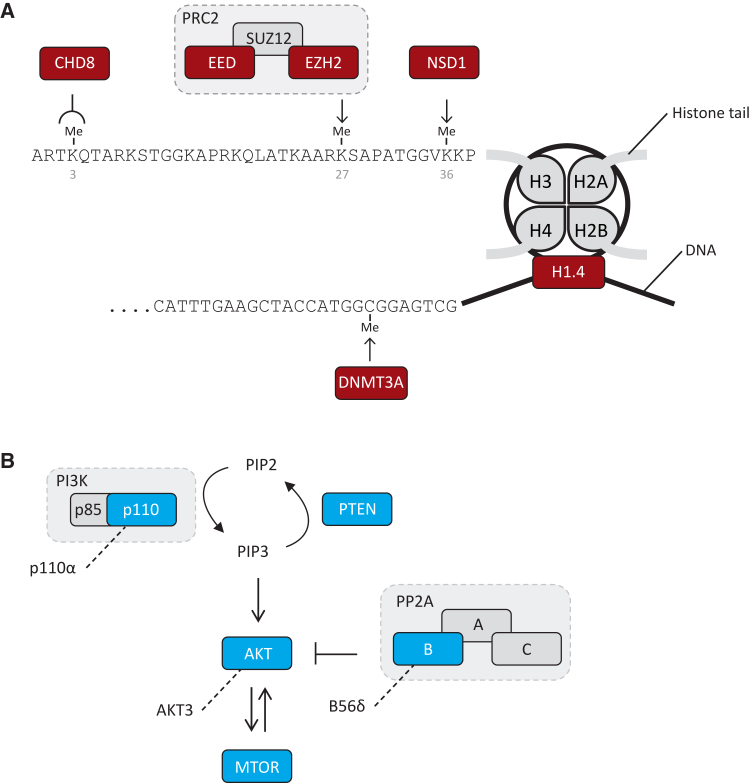
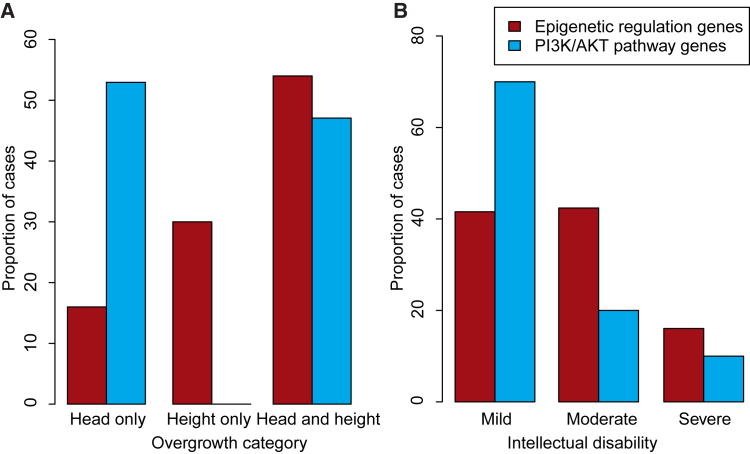
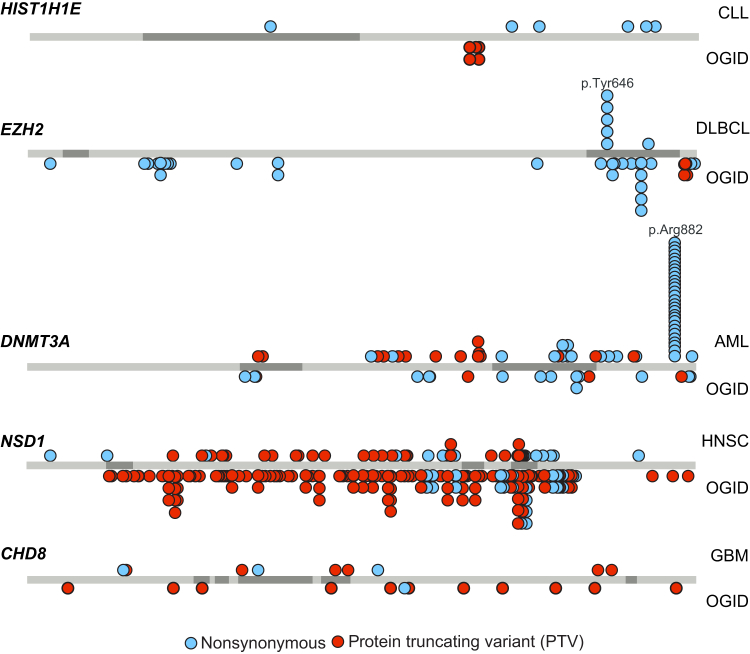
To explore the genetic architecture of human overgrowth syndromes and human growth control, we performed experimental and bioinformatic analyses of 710 individuals with overgrowth (height and/or head circumference ≥+2 SD) and intellectual disability (OGID). We identified a causal mutation in 1 of 14 genes in 50% (353/710). This includes HIST1H1E, encoding histone H1.4, which has not been associated with a developmental disorder previously. The pathogenic HIST1H1E mutations are predicted to result in a product that is less effective in neutralizing negatively charged linker DNA because it has a reduced net charge, and in DNA binding and protein-protein interactions because key residues are truncated. Functional network analyses demonstrated that epigenetic regulation is a prominent biological process dysregulated in individuals with OGID. Mutations in six epigenetic regulation genes-NSD1, EZH2, DNMT3A, CHD8, HIST1H1E, and EED-accounted for 44% of individuals (311/710). There was significant overlap between the 14 genes involved in OGID and 611 genes in regions identified in GWASs to be associated with height (p = 6.84 × 10-8), suggesting that a common variation impacting function of genes involved in OGID influences height at a population level. Increased cellular growth is a hallmark of cancer and there was striking overlap between the genes involved in OGID and 260 somatically mutated cancer driver genes (p = 1.75 × 10-14). However, the mutation spectra of genes involved in OGID and cancer differ, suggesting complex genotype-phenotype relationships. These data reveal insights into the genetic control of human growth and demonstrate that exome sequencing in OGID has a high diagnostic yield.
Keywords: EZH2; HIST1H1E; NSD1; epigenetic regulation; exome sequencing; intellectual disability; overgrowth syndrome; sotos syndrome; weaver syndrome.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Stocker H., Hafen E. Genetic control of cell size. Curr. Opin. Genet. Dev. 2000;10:529–535. - PubMed
-
- Saucedo L.J., Edgar B.A. Why size matters: altering cell size. Curr. Opin. Genet. Dev. 2002;12:565–571. - PubMed
-
- Tatton-Brown K., Weksberg R. Molecular mechanisms of childhood overgrowth. Am. J. Med. Genet. C. Semin. Med. Genet. 2013;163C:71–75. - PubMed
-
- Tatton-Brown K., Murray A., Hanks S., Douglas J., Armstrong R., Banka S., Bird L.M., Clericuzio C.L., Cormier-Daire V., Cushing T., Childhood Overgrowth Consortium Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. Am. J. Med. Genet. A. 2013;161A:2972–2980. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
