

Widespread Allelic Heterogeneity in Complex Traits
- PMID: 28475861
- PMCID: PMC5420356
- DOI: 10.1016/j.ajhg.2017.04.005
Widespread Allelic Heterogeneity in Complex Traits
Abstract
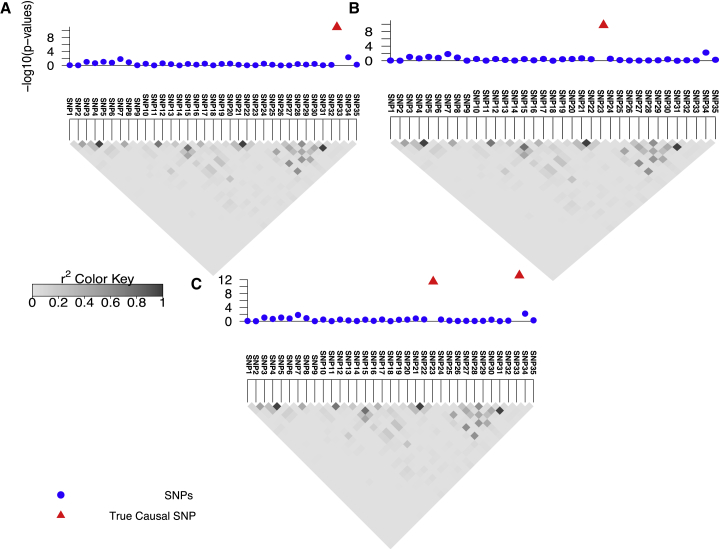
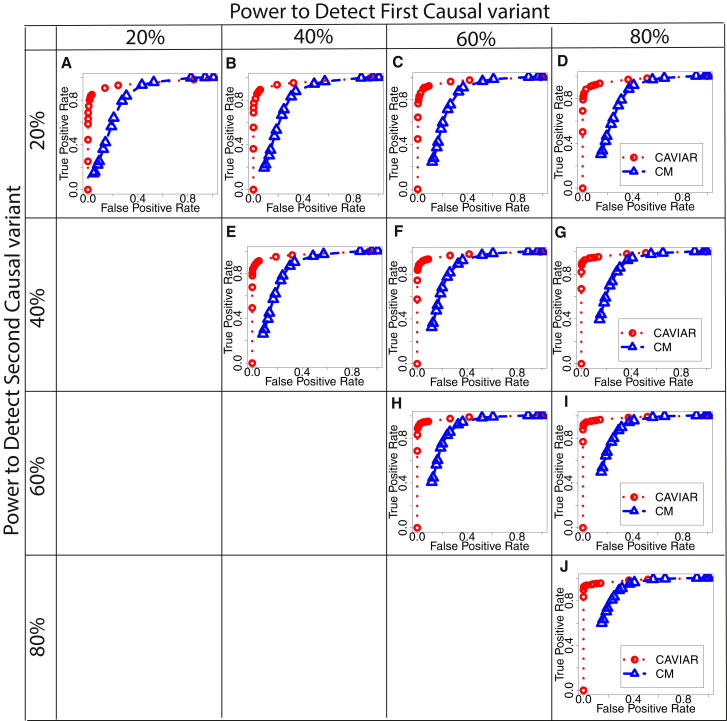
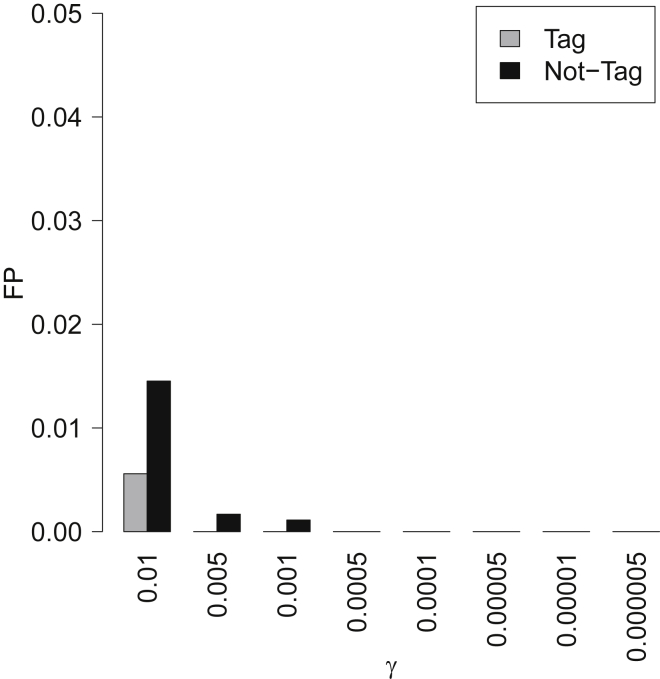
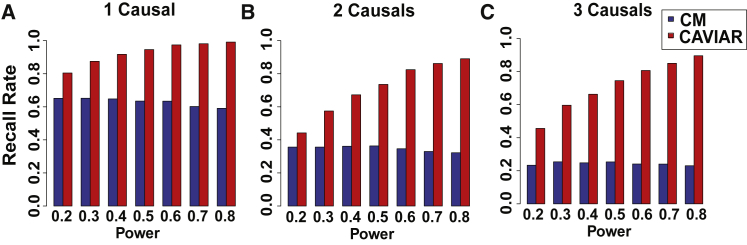
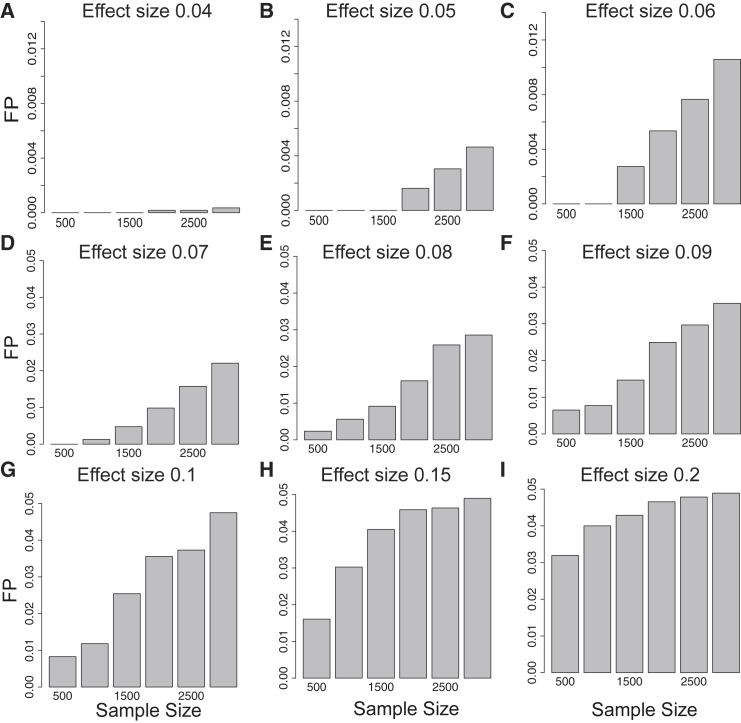
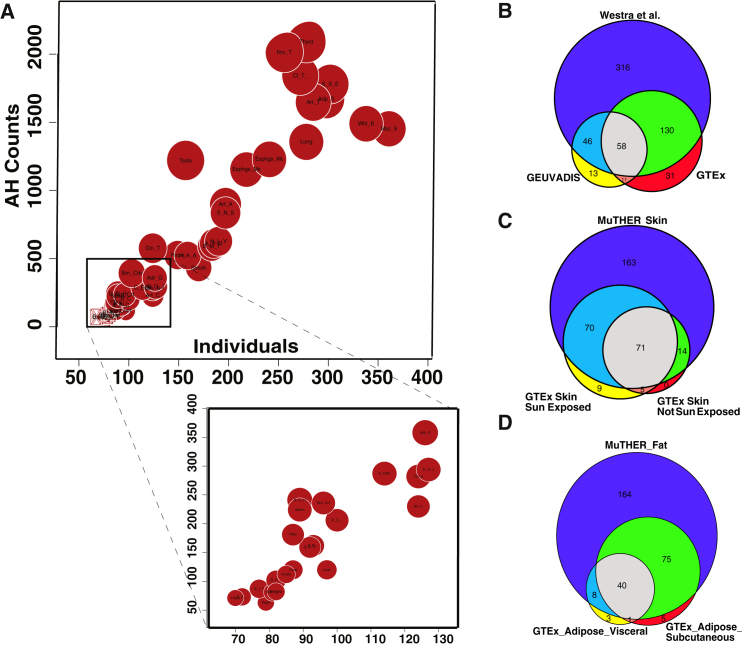
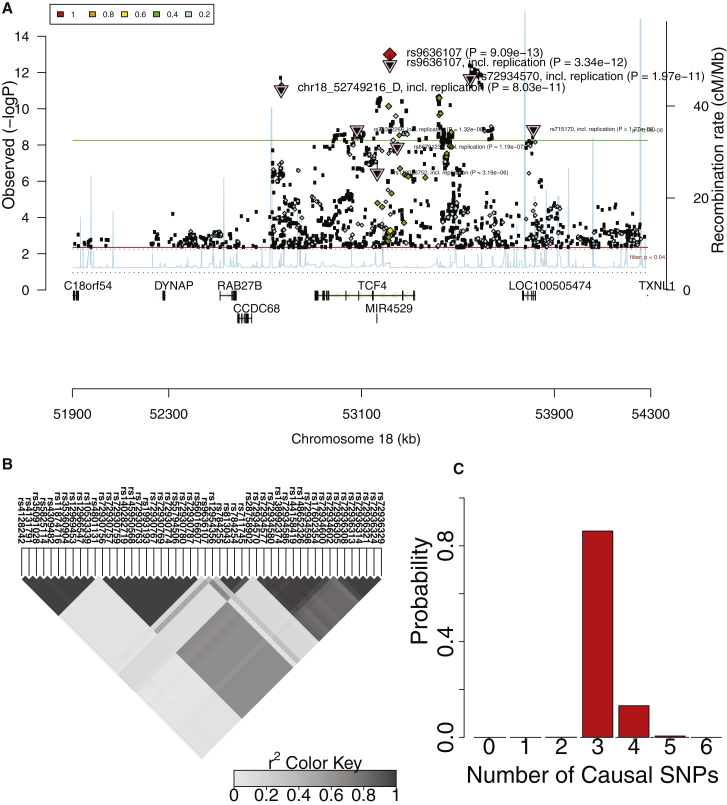
Recent successes in genome-wide association studies (GWASs) make it possible to address important questions about the genetic architecture of complex traits, such as allele frequency and effect size. One lesser-known aspect of complex traits is the extent of allelic heterogeneity (AH) arising from multiple causal variants at a locus. We developed a computational method to infer the probability of AH and applied it to three GWASs and four expression quantitative trait loci (eQTL) datasets. We identified a total of 4,152 loci with strong evidence of AH. The proportion of all loci with identified AH is 4%-23% in eQTLs, 35% in GWASs of high-density lipoprotein (HDL), and 23% in GWASs of schizophrenia. For eQTLs, we observed a strong correlation between sample size and the proportion of loci with AH (R2 = 0.85, p = 2.2 × 10-16), indicating that statistical power prevents identification of AH in other loci. Understanding the extent of AH may guide the development of new methods for fine mapping and association mapping of complex traits.
Keywords: allelic heterogeneity; causal variants; complex traits; eQTL; gene expression.
Copyright © 2017. Published by Elsevier Inc.
Figures
References
-
- Ripke S., Neale B.M., Corvin A., Walters J.T.R., Farh K.-H., Holmans P.A., Lee P., Bulik-Sullivan B., Collier D.A., Huang H., Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. - PMC - PubMed
-
- Barrett J.C., Clayton D.G., Concannon P., Akolkar B., Cooper J.D., Erlich H.A., Julier C., Morahan G., Nerup J., Nierras C., Type 1 Diabetes Genetics Consortium Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009;41:703–707. - PMC - PubMed
MeSH terms
Grants and funding
- R01 ES021801/ES/NIEHS NIH HHS/United States
- R01 MH101782/MH/NIMH NIH HHS/United States
- T32 CA201160/CA/NCI NIH HHS/United States
- K25 HL080079/HL/NHLBI NIH HHS/United States
- P30 NS062691/NS/NINDS NIH HHS/United States
- P01 HL028481/HL/NHLBI NIH HHS/United States
- U01 DA024417/DA/NIDA NIH HHS/United States
- U54 EB020403/EB/NIBIB NIH HHS/United States
- K99 MH113823/MH/NIMH NIH HHS/United States
- R01 ES022282/ES/NIEHS NIH HHS/United States
- R00 GM111744/GM/NIGMS NIH HHS/United States
- P01 HL030568/HL/NHLBI NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- R01 GM083198/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
