

Protein kinase C as a tumor suppressor
- PMID: 28476658
- PMCID: PMC5668200
- DOI: 10.1016/j.semcancer.2017.04.017
Protein kinase C as a tumor suppressor
Abstract
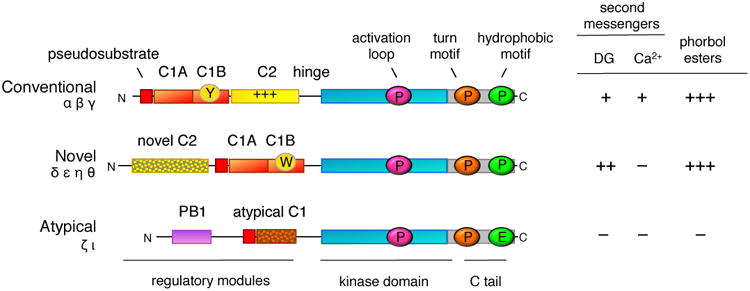
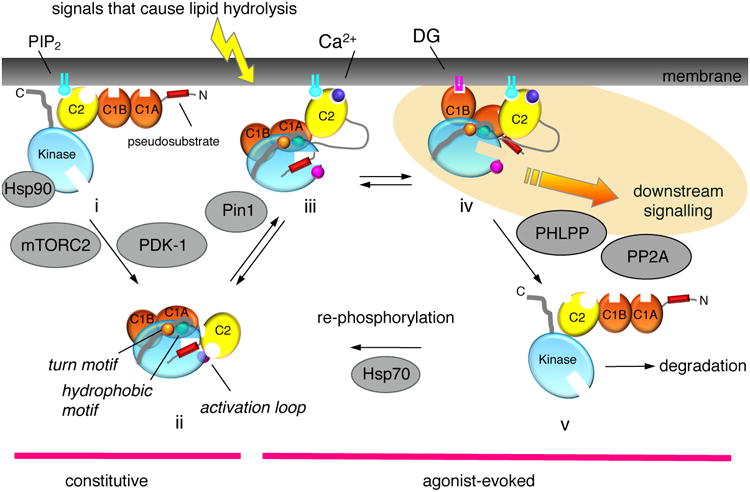
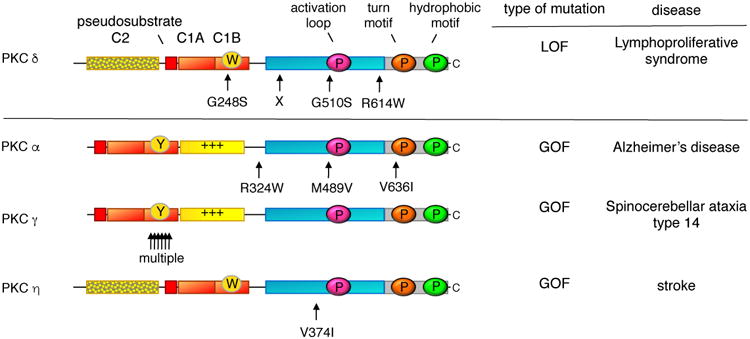
Protein kinase C (PKC) has historically been considered an oncoprotein. This stems in large part from the discovery in the early 1980s that PKC is directly activated by tumor-promoting phorbol esters. Yet three decades of clinical trials using PKC inhibitors in cancer therapies not only failed, but in some cases worsened patient outcome. Why has targeting PKC in cancer eluded successful therapies? Recent studies looking at the disease for insight provide an explanation: cancer-associated mutations in PKC are generally loss-of-function (LOF), supporting an unexpected function as tumor suppressors. And, contrasting with LOF mutations in cancer, germline mutations that enhance the activity of some PKC isozymes are associated with degenerative diseases such as Alzheimer's disease. This review provides a background on the diverse mechanisms that ensure PKC is only active when, where, and for the appropriate duration needed and summarizes recent findings converging on a paradigm reversal: PKC family members generally function by suppressing, rather than promoting, survival signaling.
Keywords: Diacylglycerol; LOF; PKC; Phorbol esters; Tumor suppressor.
Copyright © 2017 Elsevier Ltd. All rights reserved.
Figures
References
-
- Takai Y, et al. Unsaturated Diacylglycerol as a Possible Messenger for the Activation of Calcium-activated, Phospholipid-dependent Protein Kinase System. Biochem Biophys Res Comm. 1979;91:1218–1224. - PubMed
-
- Hokin MR, Hokin LE. Enzyme Secretion and the Incorporation of P32 into Phospholipids of Pancreas Slices. J Biol Chem. 1953;203:967–977. - PubMed
-
- Carrasco S, Merida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. 2007;32(1):27–36. - PubMed
-
- Manning G, et al. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–34. - PubMed
-
- Levin DE, et al. A Candidate Protein Kinase C Gene, PKC1, Is Required for the S. cerevisiae Cell Cycle. Cell. 1990;62:213–224. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
