

Self-adjusting synthetic gene circuit for correcting insulin resistance
- PMID: 28480128
- PMCID: PMC5412959
- DOI: 10.1038/s41551-016-0005
Self-adjusting synthetic gene circuit for correcting insulin resistance
Abstract
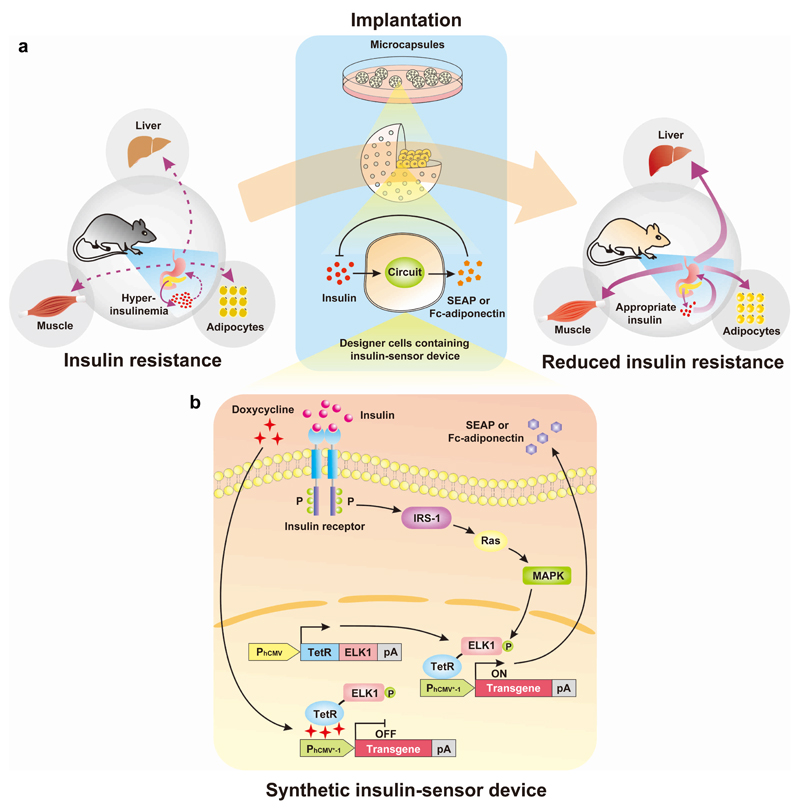
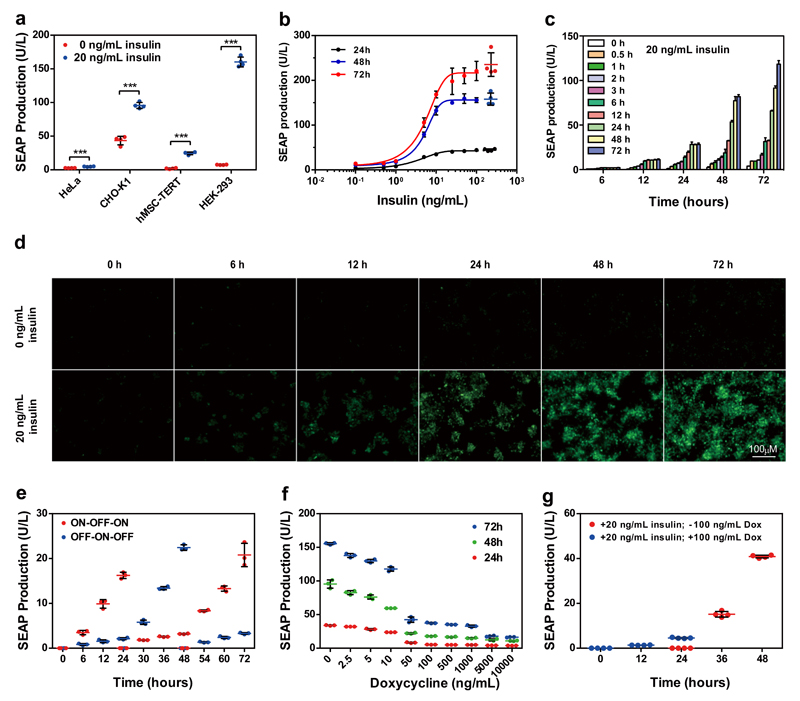
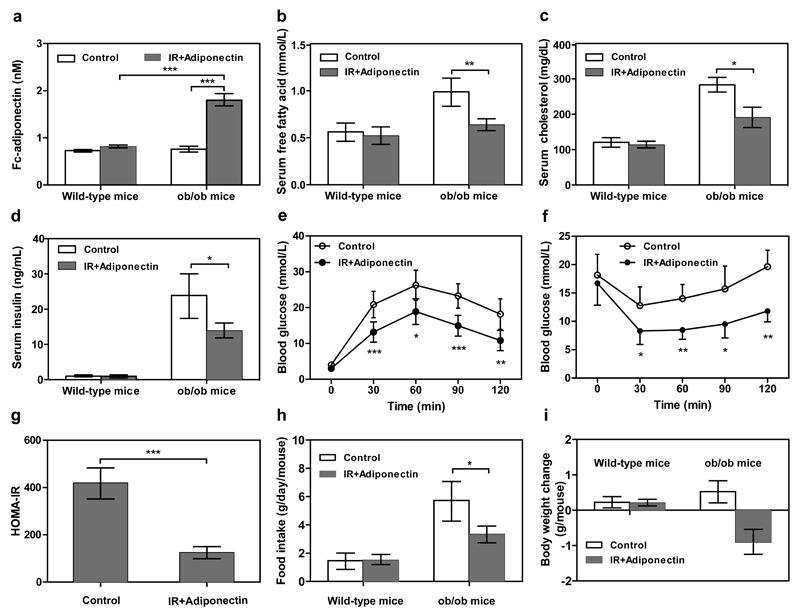
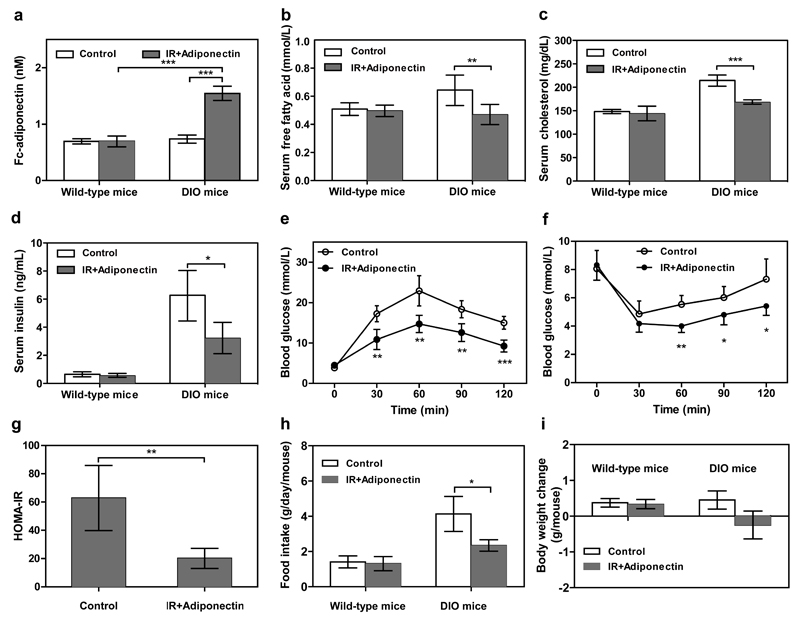
By using tools from synthetic biology, sophisticated genetic devices can be assembled to reprogram mammalian cell activities. Here, we demonstrate that a self-adjusting synthetic gene circuit can be designed to sense and reverse the insulin-resistance syndrome in different mouse models. By functionally rewiring the mitogen-activated protein kinase (MAPK) signalling pathway to produce MAPK-mediated activation of the hybrid transcription factor TetR-ELK1, we assembled a synthetic insulin-sensitive transcription-control device that self-sufficiently distinguished between physiological and increased blood insulin levels and correspondingly fine-tuned the reversible expression of therapeutic transgenes from synthetic TetR-ELK1-specific promoters. In acute experimental hyperinsulinemia, the synthetic insulin-sensing designer circuit reversed the insulin-resistance syndrome by coordinating expression of the insulin-sensitizing compound adiponectin. Engineering synthetic gene circuits to sense pathologic markers and coordinate the expression of therapeutic transgenes may provide opportunities for future gene- and cell-based treatments of multifactorial metabolic disorders.
Conflict of interest statement
Competing financial interests The authors declare no competing financial interests.
Figures
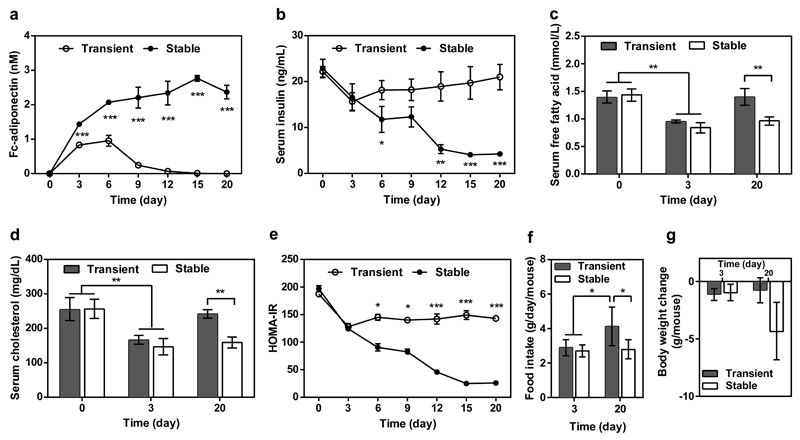
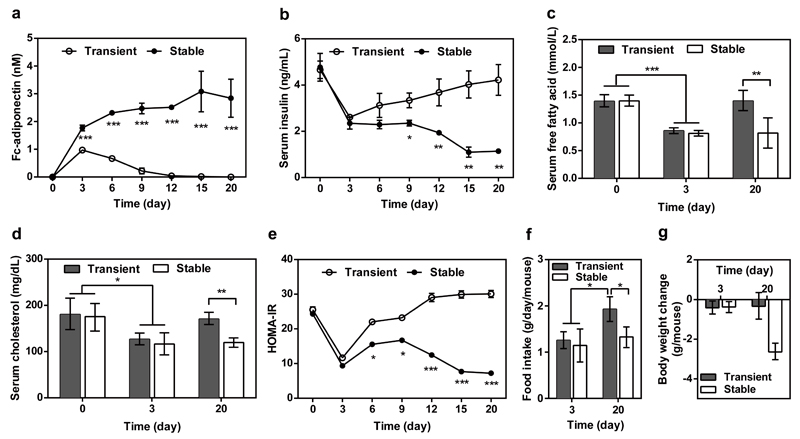
Comment in
-
Synthetic biology: Designer cells tackle diabetes.Nat Rev Mol Cell Biol. 2017 Feb;18(2):69. doi: 10.1038/nrm.2016.175. Epub 2017 Jan 5. Nat Rev Mol Cell Biol. 2017. PMID: 28053346 No abstract available.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
