

Recurrent noncoding regulatory mutations in pancreatic ductal adenocarcinoma
- PMID: 28481342
- PMCID: PMC5659388
- DOI: 10.1038/ng.3861
Recurrent noncoding regulatory mutations in pancreatic ductal adenocarcinoma
Abstract
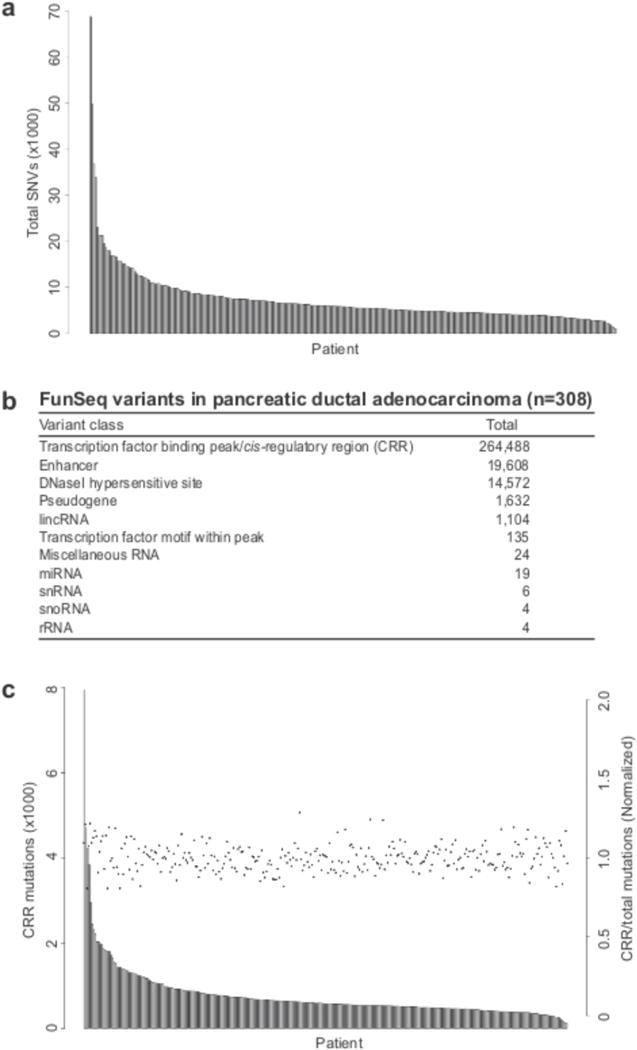
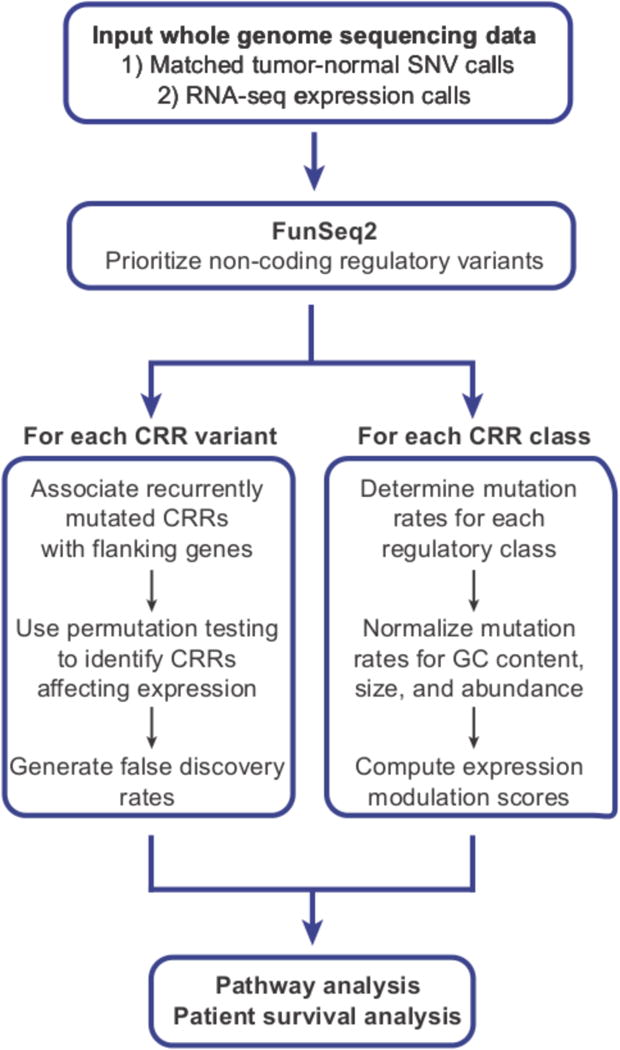
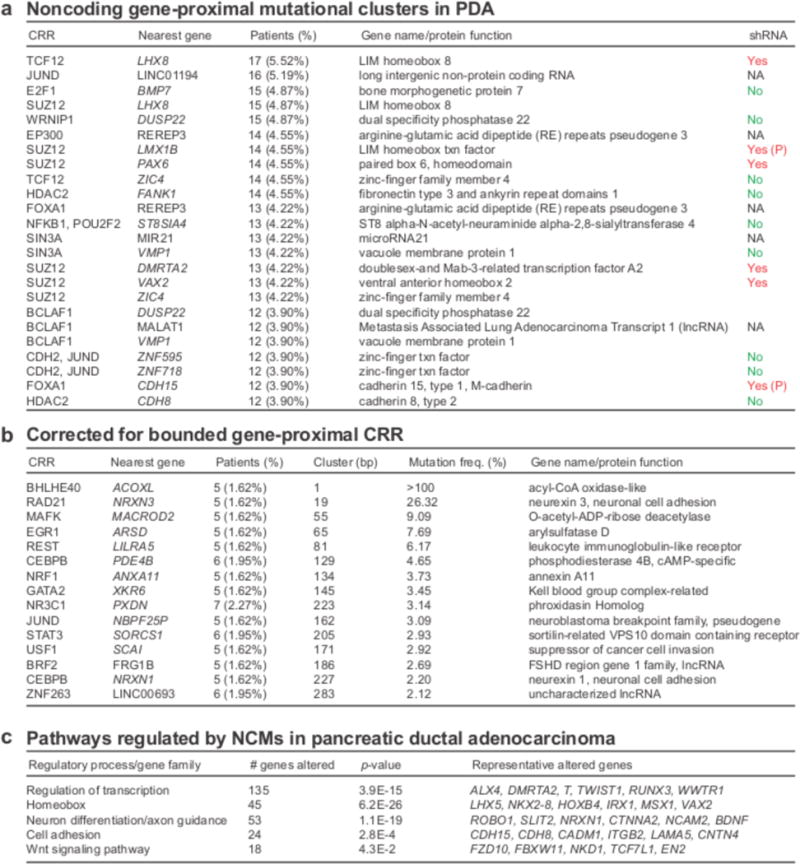
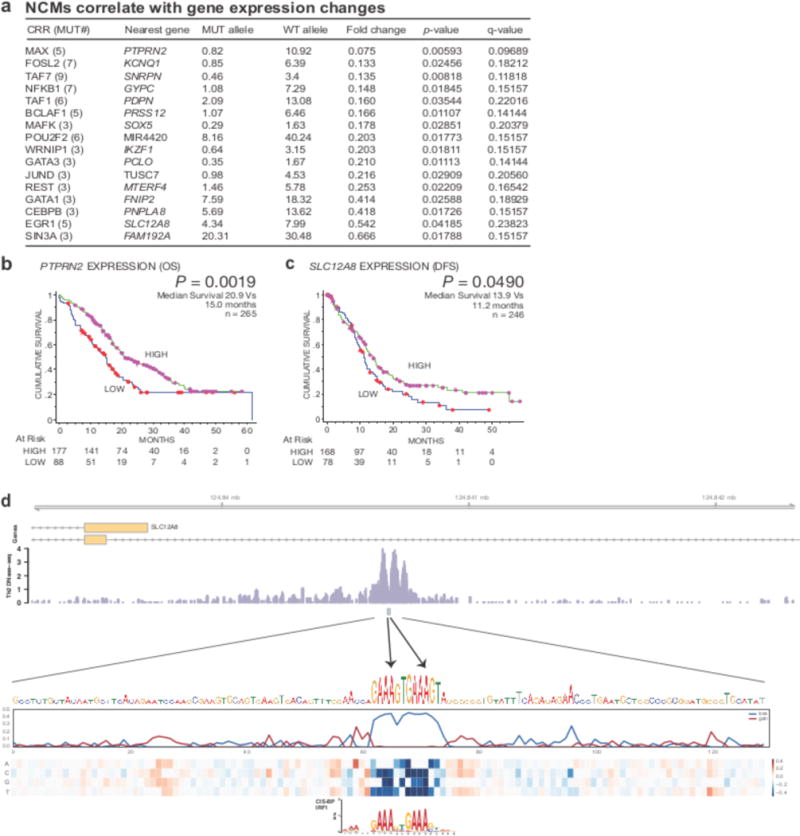
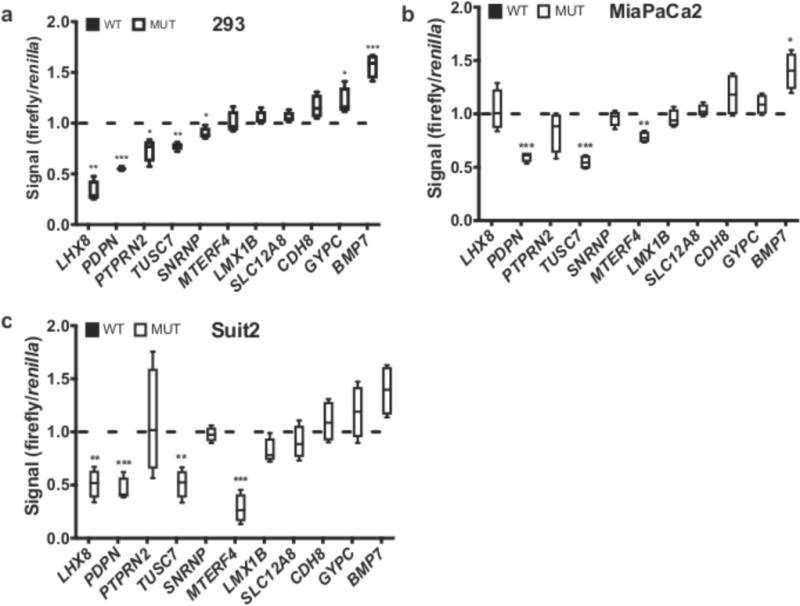
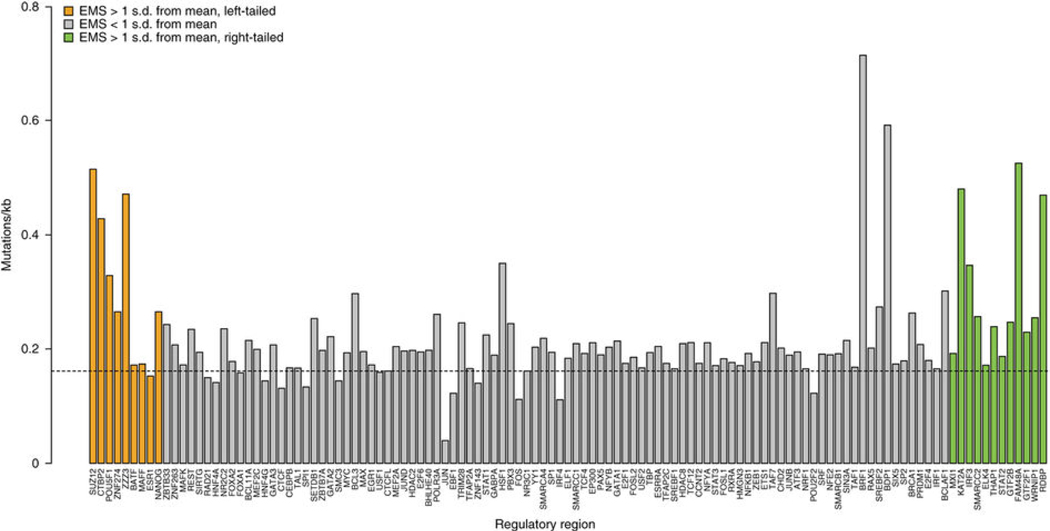
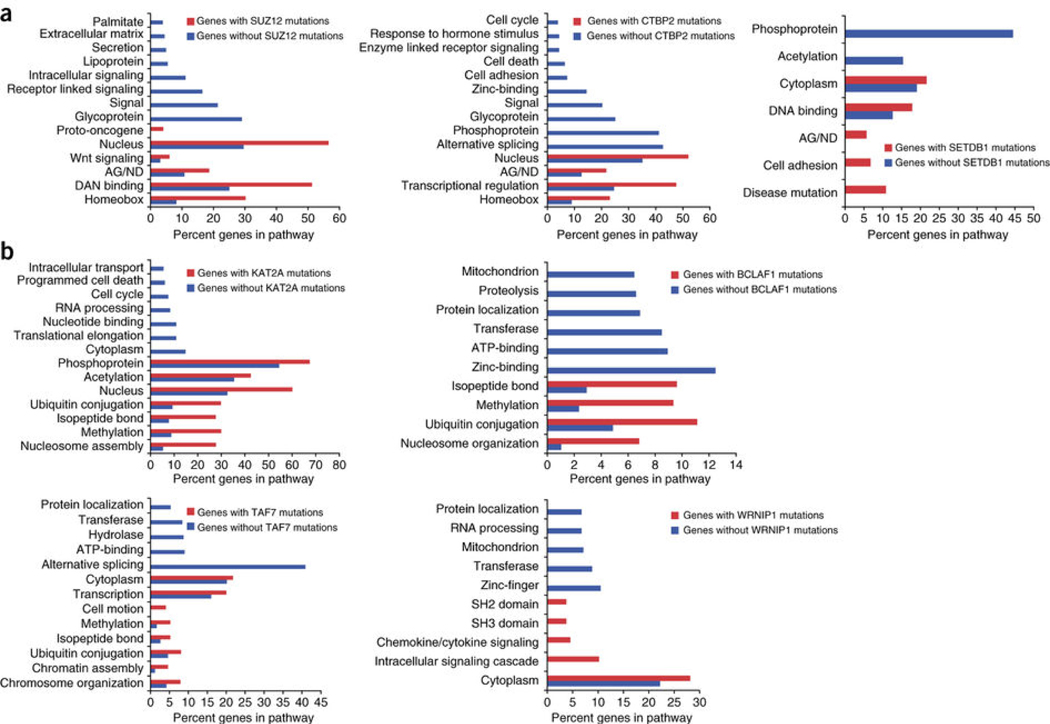
The contributions of coding mutations to tumorigenesis are relatively well known; however, little is known about somatic alterations in noncoding DNA. Here we describe GECCO (Genomic Enrichment Computational Clustering Operation) to analyze somatic noncoding alterations in 308 pancreatic ductal adenocarcinomas (PDAs) and identify commonly mutated regulatory regions. We find recurrent noncoding mutations to be enriched in PDA pathways, including axon guidance and cell adhesion, and newly identified processes, including transcription and homeobox genes. We identified mutations in protein binding sites correlating with differential expression of proximal genes and experimentally validated effects of mutations on expression. We developed an expression modulation score that quantifies the strength of gene regulation imposed by each class of regulatory elements, and found the strongest elements were most frequently mutated, suggesting a selective advantage. Our detailed single-cancer analysis of noncoding alterations identifies regulatory mutations as candidates for diagnostic and prognostic markers, and suggests new mechanisms for tumor evolution.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
